Genetically low vitamin D concentrations and increased mortality: mendelian randomisation analysis in three large cohorts
BMJ 2014; 349 doi: https://doi.org/10.1136/bmj.g6330 (Published 18 November 2014) Cite this as: BMJ 2014;349:g6330
- Shoaib Afzal, registrar12,
- Peter Brøndum-Jacobsen, registrar12,
- Stig E Bojesen, professor123,
- Børge G Nordestgaard, professor123
- 1Department of Clinical Biochemistry and Copenhagen General Population Study, Herlev Hospital, Copenhagen University Hospital, DK-2730 Herlev, Denmark
- 2Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Copenhagen City Heart Study, Frederiksberg Hospital, Copenhagen University Hospital, Frederiksberg, Denmark
- Correspondence to: B G Nordestgaard Boerge.Nordestgaard{at}regionh.dk
- Accepted 3 October 2014
Abstract
Objective To test the hypothesis that genetically low 25-hydroxyvitamin D concentrations are associated with increased mortality.
Design Mendelian randomisation analysis.
Setting Copenhagen City Heart Study, Copenhagen General Population Study, and Copenhagen Ischemic Heart Disease Study.
Participants 95 766 white participants of Danish descent from three cohorts, with median follow-up times of 19.1, 5.8, and 7.9 years, genotyped for genetic variants in DHCR7 and CYP2R1 affecting plasma 25-hydroxyvitamin D concentrations; 35 334 also had plasma 25-hydroxyvitamin D measurements. Participants were followed from study entry through 2013, during which time 10 349 died.
Main outcome measures All cause mortality and cause specific mortality, adjusted for common risk factors for all cause mortality based on the World Health Organization’s global health status.
Results The multivariable adjusted hazard ratios for a 20 nmol/L lower plasma 25-hydroxyvitamin D concentration were 1.19 (95% confidence interval 1.14 to 1.25) for all cause mortality, 1.18 (1.09 to 1.28) for cardiovascular mortality, 1.12 (1.03 to 1.22) for cancer mortality, and 1.27 (1.15 to 1.40) for other mortality. Each increase in DHCR7/CYP2R1 allele score was associated with a 1.9 nmol/L lower plasma 25-hydroxyvitamin D concentration and with increased all cause, cancer, and other mortality but not with cardiovascular mortality. The odds ratio for a genetically determined 20 nmol/L lower plasma 25-hydroxyvitamin D concentration was 1.30 (1.05 to 1.61) for all cause mortality, with a corresponding observational multivariable adjusted odds ratio of 1.21 (1.11 to 1.31). Corresponding genetic and observational odds ratios were 0.77 (0.55 to 1.08) and 1.13 (1.03 to 1.24) for cardiovascular mortality, 1.43 (1.02 to 1.99) and 1.10 (1.02 to 1.19) for cancer mortality, and 1.44 (1.01 to 2.04) and 1.17 (1.06 to 1.29) for other mortality. The results were robust in sensitivity analyses.
Conclusions Genetically low 25-hydroxyvitamin D concentrations were associated with increased all cause mortality, cancer mortality, and other mortality but not with increased cardiovascular mortality. These findings are compatible with the notion that genetically low 25-hydroxyvitamin D concentrations may be causally associated with cancer and other mortality but also suggest that the observational association with cardiovascular mortality could be the result of confounding.
Introduction
Observational studies have indicated that low vitamin D concentrations are associated with increased all cause, cardiovascular, and cancer mortality.1 2 In contrast, randomised intervention trials have not clearly documented reduced mortality after vitamin D supplementation.3 Whether low vitamin D concentrations are a cause of increased mortality or simply a consequence of poor health is thus unclear. This is an important question, as millions of people worldwide are regularly taking vitamin D supplements, presumably with the aim of preventing diseases and hopefully living longer.
Genetic studies are largely free of confounding, as the random assortment of genes that occurs during gamete formation secures an equal distribution of confounding factors among different genotypes; they can therefore be used for assessing whether genetically affected risk factors are related to clinical outcomes, also termed the mendelian randomisation approach.4 5 Genetic variants in DHCR7 and CYP2R1 that specifically decrease plasma 25-hydroxyvitamin D concentration,6 7 8 usually used to asses vitamin D status, provide a framework to assess the consequences (if any) of lifelong low 25-hydroxyvitamin D concentrations independent of other risk factors. However, three key assumptions underlie the mendelian randomisation approach: the genotypes are randomised; the genetic variants considered as instruments affect the outcome only by modifying the biomarker—that is, these variants have no pleiotropic effects on the outcome; and the genotype is independent of measured and unmeasured confounders. Although all are not directly testable, we will consider these assumptions to ensure the validity of our analyses.
The usefulness of genetic studies into modifiable risk factors is that they can often predict possible therapeutic targets to decrease morbidity and mortality. For example, genetic variants associated with increased low density lipoprotein cholesterol also increase the risk of cardiovascular disease and mortality in accordance with observational studies,9 and randomised intervention trials have proved that a decrease in low density lipoprotein cholesterol reduces cardiovascular disease and all cause mortality.10 Conversely, genetic variants associated with decreased high density lipoprotein cholesterol were not associated with increased risk of cardiovascular disease,11 12 13 14 as would be expected from observational studies,15 and randomised intervention trials have confirmed that increasing high density lipoprotein cholesterol does not decrease the risk of cardiovascular disease.16 17 18 19 Thus, genetic studies may be used to identify risk factors causally associated with an outcome, such as low density lipoprotein cholesterol, or to identify risk factors that are probably not causally related to an outcome, such as high density lipoprotein cholesterol, even though the association seems to be strong in observational studies.
We tested the hypothesis that genetically low 25-hydroxyvitamin D concentrations are associated with increased mortality. We tested firstly whether 25-hydroxyvitamin D concentrations were associated with all cause and cause specific mortality, secondly and thirdly whether the selected genotypes were associated with plasma 25-hydroxyvitamin D concentrations and with all cause and cause specific mortality,6 7 8 and finally whether the selected genotypes were associated with increased all cause and cause specific mortality consistent with their effect on plasma 25-hydroxyvitamin D concentrations by using instrumental variable analysis.5
Methods
Participants
The Copenhagen City Heart Study was started in 1976-78, with follow-up examinations in 1981-83, 1991-94, and 2001-03.20 21 People aged 20-100 were randomly invited from the national Danish Central Person Register to reflect the Danish general population. In observational analyses, we included 9902 participants with plasma 25-hydroxyvitamin D measurements from the 1981-83 examination; in genetic analyses, we included 9763 participants with all genotypes from the 1991-94 and/or 2001-03 examinations. Of these, 5460 participants had both 25-hydroxyvitamin D measurements and genotype data available.
The Copenhagen General Population Study was started in 2003 with ongoing enrolment and with participants recruited as for the Copenhagen City Heart Study.20 21 In observational analyses, we included 25 432 participants with plasma 25-hydroxyvitamin D measurements; in genetic analyses, we included 79 640 participants with all genotypes. Of these, 25 332 had both 25-hydroxyvitamin D plasma measurements and genotype data available.
The Copenhagen Ischemic Heart Disease Study consists of patients from the Copenhagen area referred for coronary angiography in 1991-2011.20 21 For genetic analyses, we included 6363 participants with all genotypes.
No participants appeared in more than one study, and all were white and of Danish descent. The three cohorts have median follow-up times of 19.1, 5.8, and 7.9 years, respectively. Supplementary figure A shows the studies included in observational and genetic studies.
Plasma 25-hydroxyvitamin D measurements
Measurements using the DiaSorin Liaison 25-hydroxyvitamin D TOTAL assay were conducted blinded to mortality and genotypic data. Copenhagen General Population Study plasma samples were collected in 2004-05 (n=12 489; stored at −80°C for about five years) and in 2009-11 (n=13 091; measured on fresh samples). Copenhagen City Heart Study plasma samples were collected in 1981-83 (n=9902; stored at −20°C for about 26 years). Assay precision was tested daily, and assay accuracy was tested using an external quality control programme (Vitamin D External Quality Assessment Scheme; DEQAS). The inter-assay coefficient of variance was 10% for a low level control (~40 nmol/L) and 8% for a high level control (~135 nmol/L). Samples for measurement were those from consecutive participants for the time periods mentioned for the Copenhagen General Population Study and all available plasma samples from the Copenhagen City Heart Study (1981-83 examination).
Genotypes
Genotyping using TaqMan assays was conducted blinded to 25-hydroxyvitamin D concentrations and mortality data. Specific genotypes for 25-hydroxyvitamin D concentrations were selected as those having the strongest, largest association in genome-wide association studies7 8; we chose genetic variants around DHCR7 (rs7944926 and rs11234027) and CYP2R1 (rs10741657 and rs12794714),6 as they are expected to influence 25-hydroxyvitamin D concentrations via either synthesis of pre-vitamin D from 7-dehydroclesterol in the skin or conversion of vitamin D to 25-hydroxyvitamin D in the liver. We deliberately did not include polymorphisms in the vitamin D binding protein, as polymorphisms in vitamin D binding protein do not associate directly with 25-hydroxyvitamin D’s biological activity,22 and we excluded other previously reported genotypes after they failed to show an association with plasma 25-hydroxyvitamin D in preliminary analyses including variants in CYP27B1 and VDR (supplementary figure B). We verified genotypes by sequencing of 48 randomly selected samples in the three cohorts. Call rates for the genotypes were more than 99% after two reruns. For DHCR7 and CYP2R1, we constructed an aggregate allele score of 0-4 as the sum of the number of 25-hydroxyvitamin D lowering alleles across the two genotypes in each gene. Furthermore, we created a DHCR7/CYP2R1 allele score by counting all alleles across the four genotypes and grouping them to achieve a comparable number of participants in each allele score group. We chose to use a simple count instead of more complex weighted scores, as it has been shown to be equally valid given similar effects of the different genetic variants.23
Potential confounders
We chose confounders on the basis of the World Health Organization’s global health status report evaluating the most common risk factors for all cause mortality and their possible association with plasma 25-hydroxyvitamin D.24 Participants reported on smoking status, cumulative tobacco consumption, alcohol consumption, intensity of leisure time physical activity, level of income, and diabetes in self reported questionnaires, reviewed together with an investigator on the day of attendance. Blood pressure (mm Hg) was measured on the day of examination, and body mass index was calculated as measured weight (kilograms) divided by measured height (meters) squared. Standard hospital assays were used to measure plasma cholesterol.
Endpoints
The Danish Civil Registration System records the date of death for all people living in Denmark. The national Danish Causes of Death Registry records rank main causes of death as well as contributing causes of death, as reported by the attending physician in general practice or at a hospital or by a physician in a forensic or pathology department.25 26 We classified causes of death according to the international classification of disease 8th or 10th revision (ICD-8 or ICD-10). We classified a death as due to cancer if one of top three ranked causes of death had a diagnosis of cancer (ICD-8 140-209, ICD-10 C00-C97), as it seemed reasonable that cardiovascular complications could be caused by cancer rather than the other way around; we classified the remaining deaths as cardiovascular if one of three ranked causes of death had a cardiovascular disease diagnosis (ICD-8 390-458, ICD-10 I00-I99); and we classified all other deaths as other death. The national Danish Causes of Death Registry lags the Danish Civil Registration System by one year, so not all deaths could be classified by cause. In the observational studies 8% (660/8535) did not have a diagnosis of cause of death, and in the genetic studies it was 16% (1703/10 349). In sensitivity analyses, we also examined other categorisations of cause specific death.
Statistical analyses
We used Stata/S.E. 12.1 for analyses. We used χ2 to evaluate Hardy-Weinberg equilibrium and Haploview to evaluate linkage disequilibrium.
To exclude influence of age, sex, season (month and year for blood sample), and study on our results, we adjusted all regressions for these variables in the studies combined to maximise statistical power; alternative analyses using age, sex, season, and study specific centiles of 25-hydroxyvitamin D to eliminate the effects of seasonal variation gave similar results. We used using a generalised Hausman test to compare the estimates from observational and genetic analyses described below. Observational analyses were adjusted for age, sex, smoking status, cumulative tobacco consumption, alcohol consumption, leisure time physical activity, systolic blood pressure, body mass index, income, diabetes, plasma cholesterol, season, and study. In observational analyses, the data were more than 99% complete for the included variables; we imputed the missing data by using multivariable chained imputation (mi impute chained) with fully conditional specification. Furthermore, we corrected observational estimates for regression dilution bias by using plasma 25-hydroxyvitamin D from 400 participants without chronic diseases who participated in the 1981-83, 1991-94, and 2001-03 examinations of the Copenhagen City Heart Study27 28; this correction helps to avoid underestimation of risk estimates but does not affect whether results are significant.
Firstly, to test whether plasma 25-hydroxyvitamin D concentrations are associated with all cause and cause specific mortality, we used Cox regression models with age as time scale and left truncation (delayed entry at examination) to estimate hazard ratios for a 20 nmol/L lower plasma 25-hydroxyvitamin D concentration. Follow-up time for each participant began at the day of blood sampling and ended at death (n=8518), emigration (n=132), or April 2013, whichever occurred first. We chose the exposure, a 20 nmol/L lower plasma concentration, on the basis of comparability to previous studies and the 25-hydroxyvitamin D distribution in our cohorts, as a 20 nmol/L decline from the 75th centile and the median corresponds to the insufficient and deficient range, respectively.
Secondly, we used Cuzick’s non-parametric trend test to assess trend across genotypes and allele scores of concentrations of 25-hydroxyvitamin D. We assessed the strengths of genotypes and allele scores as instruments by using the F statistic, where F>10 indicates sufficient strength to carry out reliable instrumental variable analyses, and R2 as a measure of variation explained by genotypes and allele scores.5
Thirdly, we examined the associations of the DHCR7/CYP2R1 allele score with all cause and cause specific mortality by using Cox regression adjusted only for age, sex, study, and year of birth, as the allele scores were randomly distributed across potential confounders. Follow-up time for each participant began at the day of blood sampling and ended at death (n=10 349), emigration (n=352), or April 2013, whichever occurred first.
Fourthly, we calculated instrumental variable estimates of genetically determined odds ratios by using the Wald-type estimator, which involves taking the ratio of the mortality allele score log odds ratio to the exposure allele score coefficient and then exponentiating to express it as an odds ratio.5 29 30 We used the delta method to derive standard errors of Wald-type instrumental variable log odds ratios.29 The adjusted 25-hydroxyvitamin D allele score coefficient comes from 30 792 participants from the general population who had both genotypic and 25-hydroxyvitamin D data. We derived observational odds ratios for comparison by using logistic regression models adjusted multivariably as described above for Cox regression. Thus, the term “genetically determined 20 nmol/L lower plasma 25-hydroxyvitamin D concentration” covers calculations involving the ratio of the association of the allele score with mortality and the allele score with plasma 25-hydroxyvitamin D scaled to 20 nmol/L.
Finally, in sensitivity analyses, we also used the competing risk proportional sub-hazard models by the method of Fine and Gray,31 in which competing risk of death was accounted for. These analyses were adjusted as in the previously mentioned Cox regression models.
Results
Plasma 25-hydroxyvitamin D concentration was associated with current smoking, cumulative tobacco consumption, leisure time physical activity, systolic blood pressure, body mass index, income, diabetes, and plasma cholesterol (table⇓, supplementary table A). In contrast, the DHCR7/CYP2R1 allele score was not associated with these potential confounders (table⇓, supplementary table B), illustrating that allele score can be used as a largely unconfounded instrument to assess the association of genetically low 25-hydroxyvitamin D concentrations with all cause and cause specific mortality. DHCR7 genotypes and CYP2R1 genotypes were not in linkage disequilibrium (R2=0%), implying that genetic variants in the two genes were completely unrelated. Within each gene, the variants each explained 49% of the variation in the other. Additionally, genetic variants in DHCR7 and CYP2R1 were not associated with concentrations of cholesterol or steroid hormones that are thought to be processed by these enzymes,6 and they were not in linkage disequilibrium with other genetic variants associated with longevity or mortality in genome-wide association studies (supplementary figure C). All genotypes were in Hardy-Weinberg equilibrium (P>0.05).
Baseline characteristics and their association with plasma 25-hydroxyvitamin D (25-(OH)D) centiles and 25-(OH)D lowering allele score. Values are median (interquartile range) unless stated other wise
Plasma 25-hydroxyvitamin D and all cause and cause specific mortality: observational estimates
A 20 nmol/L lower plasma 25-hydroxyvitamin D concentration was associated with all cause and cause specific mortality, both in the two studies separately and in the pooled analyses (fig 1⇓). The multivariable adjusted hazard ratios in pooled analyses were 1.19 (95% confidence interval, 1.14 to 1.25) for all cause mortality, 1.18 (1.09 to 1.28) for cardiovascular mortality, 1.12 (1.03 to 1.22) for cancer mortality, and 1.27 (1.15 to 1.40) for other mortality.

Fig 1 Association of plasma 25-hydroxyvitamin D concentrations with all cause and cause specific mortality in general population. Analysis was by Cox regression adjusted for age, sex, smoking status, cumulative tobacco consumption, alcohol consumption, leisure time physical activity, systolic blood pressure, body mass index, income, diabetes, plasma cholesterol, season (month and year of blood sample), and study (in pooled analyses). Follow-up was up to 32 years in Copenhagen City Heart Study (CCHS) and up to 9.4 years in Copenhagen General Population Study (CGPS). 25-(OH)D=25-hydroxyvitamin D
{kind=link}
Genotype and plasma 25-hydroxyvitamin D
Plasma 25-hydroxyvitamin D concentrations were 4.6 (95% confidence interval 3.3 to 5.9) nmol/L lower for four versus no variant alleles in DHCR7 and 6.1 (5.2 to 7.0) nmol/L lower for four versus no variant alleles in CYP2R1. Combining genotypes by 25-hydroxyvitamin D lowering variant alleles showed an 8.4 (7.4 to 9.5) nmol/L decrease for 6-8 versus 0-1 variant alleles in the DHCR7/CYP2R1 allele score. Each increase in DHCR7/CYP2R1 allele score was associated with a 1.9 (1.7 to 2.1) nmol/L decrease in plasma 25-hydroxyvitamin D concentration. Figure 2⇓ shows the strength of the genotypes and allele scores as instruments and the variation in plasma 25-hydroxyvitamin D concentration explained by these instruments.

Fig 2 Concentrations of 25-hydroxyvitamin D adjusted for age, sex, season, and study according to genotypes and allele scores used as instrumental variables in genetic analyses. Columns show mean concentrations with 95% confidence intervals, P values are for trend across genotypes and allele scores, F test is for statistical strength of instrument, and R2 is measure of explained variation. 25-hydroxyvitamin D analyses are based on 30 792 participants from general population (Copenhagen City Heart Study and Copenhagen General Population Study combined), where both genotype and 25-hydroxyvitamin D were measured
{kind=link}
Allele score and all cause and cause specific mortality
The hazard ratio per one DHCR7/CYP2R1 allele score increase was 1.02 (1.00 to 1.03; P=0.03) for all cause mortality, 0.98 (0.96 to 1.01) for cardiovascular mortality, 1.03 (1.00 to 1.06; P=0.04) for cancer mortality, and 1.03 (1.00 to 1.06; P=0.07) for other mortality (fig 3⇓).

Fig 3 All cause and cause specific mortality according to DHCR7/CYP2R1 allele score. Analyses were carried out using logistic regression adjusted for age, year of birth, sex, and study (only in pooled population). CCHS=Copenhagen City Heart Study; CGPS=Copenhagen General Population Study; CIHDS=Copenhagen Ischemic Heart Disease Study
{kind=link}
Plasma 25-hydroxyvitamin D and all cause and cause specific mortality: instrumental variable estimates
The odds ratio for a genetically determined 20 nmol/L lower plasma 25-hydroxyvitamin D concentration was 1.30 (1.05 to 1.61) for all cause mortality, with a corresponding observational multivariable adjusted odds ratio of 1.21 (1.11 to 1.31) (fig 4⇓). Corresponding genetic and observational odds ratios were 0.77 (0.55 to 1.08) and 1.13 (1.03 to 1.24) for cardiovascular mortality, 1.43 (1.02 to 1.99) and 1.10 (1.02 to 1.19) for cancer mortality, and 1.44 (1.01 to 2.04) and 1.17 (1.06 to 1.29) for other mortality.
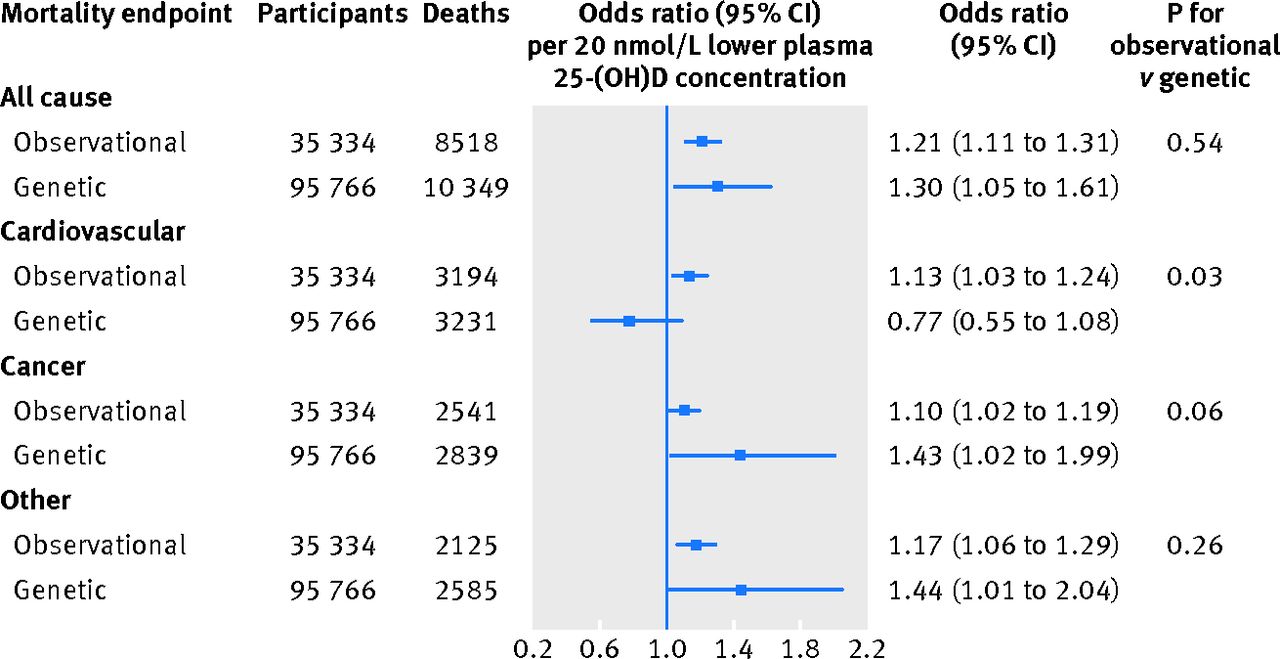
Fig 4 Observational and genetic risk estimates for all cause and cause specific mortality for 20 nmol/L lower 25-hydroxyvitamin D concentrations. Observational estimates were by logistic regression and genetic estimates by instrumental variable analyses. Observational analyses were adjusted for age, sex, smoking status, cumulative tobacco consumption, alcohol consumption, leisure time physical activity, systolic blood pressure, body mass index, income, diabetes, plasma cholesterol, season (month and year of blood sample), and study. Genetic analyses were adjusted for age, year of birth, sex, and study. Observational estimates were based on participants from Copenhagen City Heart Study and Copenhagen General Population Study combined; genetic estimates were based on participants from Copenhagen City Heart Study, Copenhagen General Population Study, and Copenhagen Ischemic Heart Disease Study combined. 25-(OH)D=25-hydroxyvitamin D
{kind=link}
Sensitivity analyses
The concentrations of plasma 25-hydroxyvitamin D varied according to season, and the association with DHCR7/CYP2R1 allele score was present in all four seasons (fig 5⇓). Using month specific centiles of plasma 25-hydroxyvitamin D to remove seasonal variation gave similar results in observational and genetic analyses (supplementary figures D and E) compared with main results (figures 1⇑ and 4⇑).

Fig 5 Seasonal variation in plasma 25-hydroxyvitamin D concentration (means with 95% CI) and association of DHCR7/CYP2R1 allele score with plasma 25-hydroxyvitamin D (25-(OH)D) according to season (effect per allele with 95% CI)
{kind=link}
In observational analyses, the associations for cause specific mortality were similar when we allowed for competing risk of death from other causes by using Fine and Gray sub-hazard regression (supplementary figure F) or when we used an alternative classification of mortality endpoints (supplementary figure G). Use of 25-hydroxyvitamin D concentration in clinical categories showed similar results with significant trends across >75 through 50.1-75, 25.1-50, and ≤25 nmol/L for all cause and cause specific mortality (supplementary figure H). Further subdivision of the mortality endpoints suggested that low plasma 25-hydroxyvitamin D concentrations strongly associated specifically with higher risk of coronary, lung cancer, and respiratory deaths (supplementary figure I). Stratified analyses according to baseline cardiovascular disease or cancer showed similar estimates for all cause, cardiovascular, and cancer mortality in relevant strata, and no interactions with baseline disease were present (supplementary figure J). Lastly, we had data on self reported vitamin or nutritional supplement intake only in the Copenhagen General Population Study, and adjustment for this variable did not affect effect estimates (supplementary figure K).
In genetic analyses, results were similar when we combined estimates by using a meta-analytic approach instead of pooling (supplementary figure L) or when we used alternative statistical models such as a Cox regression with limited follow-up corresponding to the longest follow-up in the Copenhagen General Population Study, logistic regression, and Fine and Gray sub-hazard regression accounting for competing risk of death from other causes (supplementary figures M and N).
In instrumental variable analyses, the results were similar when we used each genotype as an instrument separately, although with lower statistical power (supplementary figure O). However, the odds ratio for a genetically determined 20 nmol/L lower plasma 25-hydroxyvitamin D concentration was 0.60 (0.40 to 0.91) for cardiovascular mortality based on the CYP2R1 allele score, with a corresponding odds ratio of 1.13 (0.63 to 2.03) for the DHCR7 allele score. Additionally, alternative instruments using only one of the DHCR7 genotypes or using only the strongest instruments in DHCR7 and CYP2R1 showed similar results (supplementary figure P). Stratified analyses for age and sex showed similar results (supplementary figures Q and R).
Lastly, we carried out analyses according to restricted follow-up from 1-32 years to evaluate the dependence of risk estimates on follow-up time. Results were similar to analyses using complete follow-up (supplementary figure S).
Discussion
The findings of this study including 95 766 participants suggest that genetically low plasma 25-hydroxyvitamin D concentrations are associated with increased all cause, cancer, and other mortality but not with cardiovascular mortality. These findings are shown here for the first time in a study with sufficient sample size to investigate the association of genetically low plasma 25-hydroxyvitamin D concentrations with increased mortality.
Interpretation of findings
These findings corresponding to a 30% reduced cancer mortality for a genetically 20 nmol/L increase in 25-hydroxyvitamin D concentration, if applicable to a randomised intervention trial setting, suggest that the ongoing Vitamin D and Omega-3 Trial (VITAL) study expecting a 50 nmol/L increase in 25-hydroxyvitamin D will be sufficiently powered to detect reduced cancer mortality.32 However, recent meta-analyses of randomised intervention trials with vitamin D supplements have found smaller effects, and a recent umbrella review of meta-analyses of randomised intervention trials found little effect on mortality endpoints.1 3 33 The mendelian randomisation approach has three important differences compared with randomised intervention trials. Firstly, the use of genetics influencing plasma 25-hydroxyvitamin D potentially captures all three sources of vitamin D supply—that is, sun exposure, diet, and supplements—rather than supplements only as used in randomised trials. Secondly, lifelong exposure to 25-hydroxyvitamin D concentrations is tested rather than time limited interventions. Lastly, randomised intervention trials cannot test the case in which irreversible damage has occurred before intervention, which will often be true of diseases with a long natural history such as cancer. Thus, the estimates from randomised intervention trials may be somewhat attenuated compared with our results, as suggested by meta-analyses.1 3 33 Nevertheless, randomised intervention trials remain the gold standard to establish the usefulness of interventions, whereas mendelian randomisation studies are geared towards exploring mechanisms. Randomised intervention trials must show a benefit before widespread vitamin D supplementation can be recommended.
Mechanistically, the association of genetically low 25-hydroxyvitamin D concentrations with mortality endpoints is somewhat supported by biological, epidemiological, and clinical evidence. Firstly, vitamin D activating enzymes and the vitamin D receptor are present in many tissues,34 35 36 and vitamin D is estimated to regulate 1-3% of all gene expression.37 38 Secondly, vitamin D has been implicated in the regulation of proliferation, differentiation, and apoptosis in several cell types.38 39 40 41 42 43 Thirdly, animal studies indicate that knockout of the vitamin D receptor may reduce lifespan and accelerate ageing.44 45 Fourthly, observational studies have implicated low vitamin D concentrations as a risk factor for a wide range of diseases from autoimmune and infectious disease to cardiovascular disease and cancers.1 Finally, a meta-analysis of randomised intervention trials indicates that vitamin D3, but not vitamin D2, supplementation may decrease all cause and cancer mortality, especially among vitamin D deficient populations.3 Taken together, current evidence thus suggests that low vitamin D concentrations could be causally associated with several mortality endpoints.
Comparison with other studies
The few genetic association studies investigating the 25-hydroxyvitamin D pathway regarding mortality endpoints have been contradictory.46 47 The association of plasma 25-hydroxyvitamin D concentration with risk of major clinical outcomes including mortality varied according to genetic variants in the vitamin D receptor.46 Another study including about 3000 participants did not find an association between DHCR7 or CYP2R1 genotypes and mortality.47 However, given the sample size and only a weak association of genotypes with 25-hydroxyvitamin D, this last study was under-powered to detect the effect sizes reported here. Nevertheless, our study documents that genetically reduced vitamin D concentrations are associated with increased all cause, cancer, and other mortality. Furthermore, recent studies suggest that the cardiovascular risk factors, high remnant cholesterol, low density lipoprotein cholesterol, and obesity may causally decrease 25-hydroxyvitamin D plasma concentrations and increase risk of cardiovascular disease.6 48 49 That is, low plasma 25-hydroxyvitamin D may be a consequence of a predisposition to cardiovascular disease rather than a cause of cardiovascular disease. This could explain why we did not see an association in mendelian randomisation analyses, despite strong associations in observational studies.1 2
An alternative explanation for the negative finding regarding cardiovascular mortality could be related to the choice of instruments in our study. We chose instruments related to synthesis, but not metabolism or activity, of 25-hydroxyvitamin D, whereas including both would be an optimal strategy as suggested previously.50 We did this because we did not find any association with plasma 25-hydroxyvitamin D concentrations in a pilot study genotyped for variants in CYP27B1 and VDR (supplementary figure A), we did not include polymorphisms in the vitamin D binding protein (GC) as these do not associate with 25-hydroxyvitamin D’s biological activity in the predictable way used in mendelian randomisation studies,22 and at the time of genotyping other variants had not been consistently associated with plasma 25-hydroxyvitamin D concentrations. However, variants in the metabolic pathways may have effects not reflected in plasma 25-hydroxyvitamin D concentrations but rather in activity of 25-hydroxyvitamin D metabolites. Thus, genotyping of variants in the metabolic pathway may help in validating our findings through another genetic model, and strictly speaking one could argue that our results relate to 25-hydroxyvitamin D synthesis only and not to 25-hydroxyvitamin D metabolism.
Lastly, we cannot rule out that the lack of association is due to lack of power. In our setting, the power to detect an odds ratio of 1.5 is 85%, and it is only 44% for an odds ratio of 1.3, for the cardiovascular mortality endpoint using the mendelian randomisation approach.51
Strengths and limitations of study
Strengths of our study include a large population size, with sufficient statistical power to detect an association of 25-hydroxyvitamin D lowering genotypes with mortality; the fact that our instruments were strong (F>>10) for instrumental variable analysis; and that the results were robust in sensitivity analyses. Furthermore, we did not detect any violations of the assumptions underlying mendelian randomisation as far as they could be tested. Also, as we included participants at random and consecutively from the general population, both for genetic and plasma 25-hydroxyvitamin D analyses, the potential for selection bias is minimal. Additionally, in northern Europe, ultraviolet B radiation from the sun is adequate for sufficient endogenous vitamin D production in the skin only during the summer months. Finally, the policy regarding food fortification with vitamin D in Denmark has been restrictive throughout the study period, and the general population of Denmark has minor intake of vitamin D through the diet.52 This sample from the Danish general population thus allows determination of the natural history of the association of vitamin D deficiency with risk of mortality.
Potential limitations of the mendelian randomisation approach include presence of genetic pleiotropy and linkage disequilibrium between the genetic variants used as instruments with other genetic variants associated with the outcome. However, as shown in results, we saw no evidence of genetic pleiotropy or linkage disequilibrium with other genetic variants associated with longevity or mortality in genome-wide association studies. Although population homogeneity eliminates population admixture as a potential confounder in our study, 25-hydroxyvitamin D concentrations are known to vary with sun exposure and skin colour, and we studied only white Danes, thus potentially limiting the generalisability of the results to other geographical regions. Nevertheless, we tried to overcome this problem indirectly. Firstly, we showed that the DHCR7/CYP2R1 allele score was associated with 25-hydroxyvitamin D concentrations across all seasons. Secondly, variables associated with higher plasma 25-hydroxyvitamin D concentration, and presumably higher sun exposure, did not interact with the DHCR7/CYP2R1 allele score on risk of mortality in the general population (data not shown), indicating that the associations may remain similar with different levels of sun exposure. However, future studies need to test this directly. The different storage conditions and delays in measurements could raise concern about potential decay of plasma 25-hydroxyvitamin D and bias in associations, but this seems unlikely for several reasons as reported previously28 53: we noticed the expected seasonal variation of 25-hydroxyvitamin D concentrations; median concentrations of plasma 25-hydroxyvitamin D across plasma samples from three different examinations on the same healthy participants with storage times of 10, 20, and 30 years were similar; previous studies have shown high stability during storage; the median concentrations observed in our studies are similar to those in comparable populations; and a low sample quality for the 25-hydroxyvitamin D measurement would tend to weaken rather than inflate an association.
Another point to consider is that we assumed a linear association of plasma 25-hydroxyvitamin D concentrations with mortality. However, the association is rather curvilinear than linear, as suggested by some of our sensitivity analyses. The observed association for the linear association thus probably underestimates the true effect estimates, because a 20 nmol/L change in 25-hydroxyvitamin D concentrations in participants with sufficient 25-hydroxyvitamin D concentrations (>50 nmol/L) may not be associated with lower mortality. Nevertheless, we used the continuous 25-hydroxyvitamin D variable because of the higher statistical power compared with a categorical variable, as low power is often a major problem in mendelian randomisation studies. Lastly, effects estimates for some genetic analyses may seem low, but they are comparable to those seen in well established causal associations, such as smoking with all cause mortality and lipids with ischaemic heart disease, using the same study populations.54 55
Conclusion
Genetically low 25-hydroxyvitamin D concentrations were associated with increased all cause mortality, cancer mortality, and other mortality but not with cardiovascular mortality. These findings are compatible with the notion that genetically low 25-hydroxyvitamin D concentrations may be causally associated with mortality due to cancer and other causes, but also suggest that the observational association with cardiovascular mortality could be the result of confounding. The clinical implication of our findings remain limited, as widespread vitamin D supplementation can be recommended only after benefit is shown in randomised intervention trials.
What is already known on this topic
-
Observational studies have suggested that lower concentrations of vitamin D are associated with increased mortality, but such associations can be difficult to interpret owing to bias and confounding
-
Genetic variants in DHCR7 and CYP2R1 have been reliably associated with circulating concentrations of 25-hydroxyvitamin D, a marker of vitamin D status
-
These variants have been proposed as an instrument to investigate causal associations between vitamin D status and clinical outcomes
What this study adds
-
Genetically low 25-hydroxyvitamin D concentrations were associated with increased all cause mortality, cancer mortality, and other mortality but not with cardiovascular mortality
-
These findings suggest that low vitamin D status may be causally associated with cancer and other non-cardiovascular mortality, a finding with potential public health implications
-
Vitamin D supplementation may not be a clinically relevant intervention in cardiovascular disease prevention
Notes
Cite this as: BMJ 2014;349:g6330
Footnotes
-
Contributors: SA and BGN conceived and coordinated the investigation. SA wrote the manuscript. SEB and PB-J were responsible for the preparation of data, and SA, PBJ, SEB, and BGN undertook revisions and contributed intellectually to the development of this paper. SA is the guarantor.
-
Funding: The Copenhagen General Population Study and Copenhagen City Heart Study are supported by the Danish Heart Foundation, Danish Medical Research Council, Copenhagen County Foundation, and Herlev Hospital, Copenhagen University Hospital. All researchers operate independently of the funding bodies.
-
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work.
-
Ethical approval: The studies were approved by the Danish ethical committees and Herlev Hospital, Copenhagen University Hospital. Participants gave written informed consent.
-
Data sharing: Additional data regarding technical details, statistical code, and derivative data are available from the principal investigator at boerge.nordestgaard{at}regionh.dk. Data access for further analyses is possible through direct collaborative agreement or through locally managed access arranged through the study’s principal investigator.
-
Transparency: The lead author (the manuscript’s guarantor) affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.