Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats

 and
and 
Abstract
:1. Introduction
1.1. Current Standard of Care for AML
1.2. AML Cell Surface Antigens
1.3. Cancer Stem Cell Hypothesis and Optimal Antigen Targets in AML
1.4. Optimal Targets in AML Therapy (CD33, CD123, CD13, CLL-1 and CD38)
1.5. Additional Targets in AML (WT-1, CD15, CD25, CD30, CD45)
2. Unconjugated Antibody Therapies
2.1. CSL360/CSL362 (Talacotuzumab)
2.2. Lintuzimab and Bl 835858
2.3. Daratumumab (Darzalex), Isatuximab
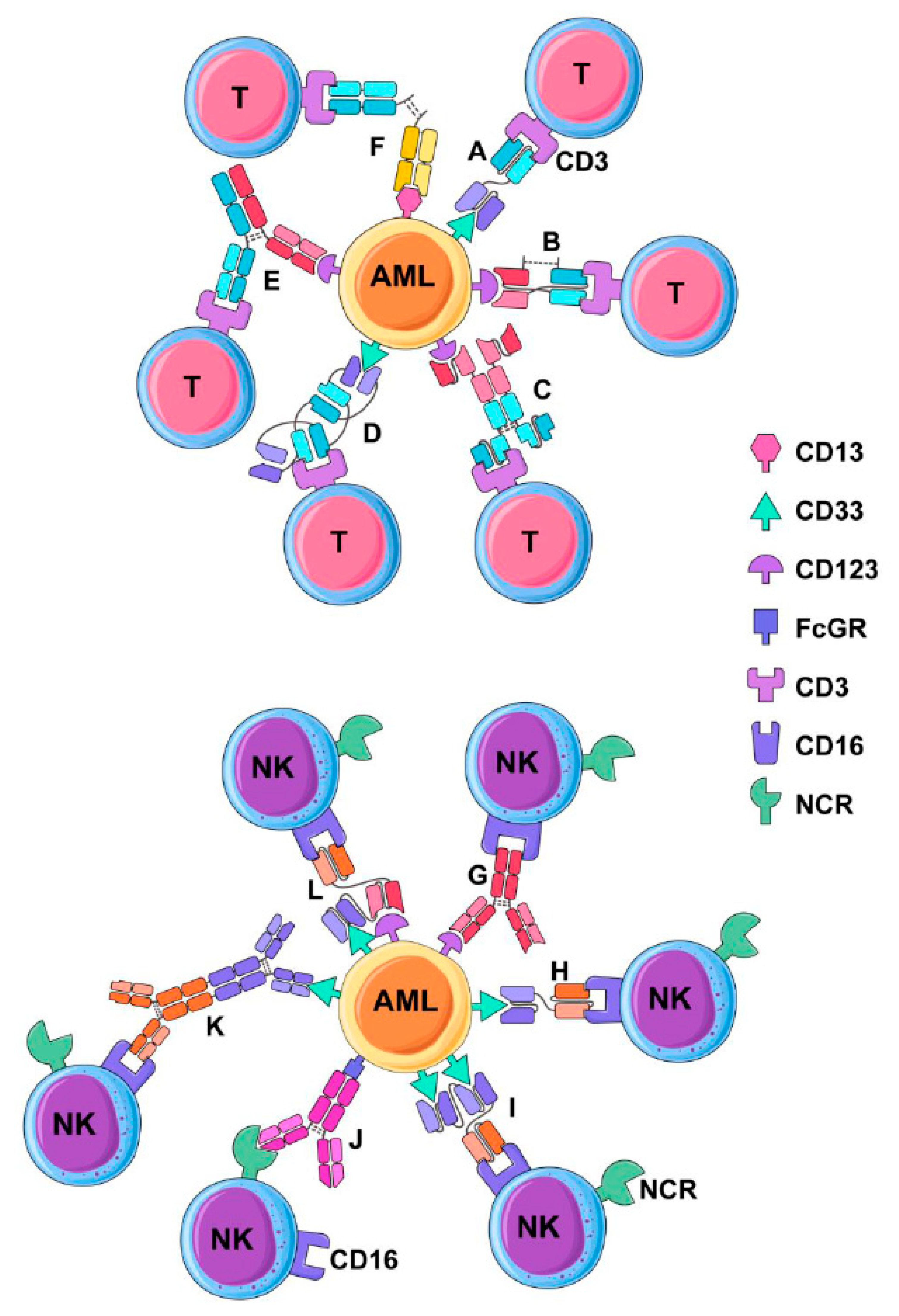
3. Multivalent Antibody Therapies
3.1. Bispecific Tandem Fragment Variable Format (BiTE, scBsTaFv)
3.2. Dual-Affinity Retargeting (DART)
3.3. Bispecific scFv Immunofusion or BIf
3.4. Bispecific Tandem Diabodies (TandAb)
3.5. Chemically Conjugated Bispecific Antibodies
3.6. Bispecific Full-Length Antibodies (Duobody and Biclonics)
3.7. BiKEs and TriKEs
4. Toxin-Conjugated Antibody Therapy for AML
4.1. Gemtuzumab Ozogamicin (GO)
4.2. Vadastuximab Talirine (SGN33A) and IMGN779
4.3. Current Clinical Trials
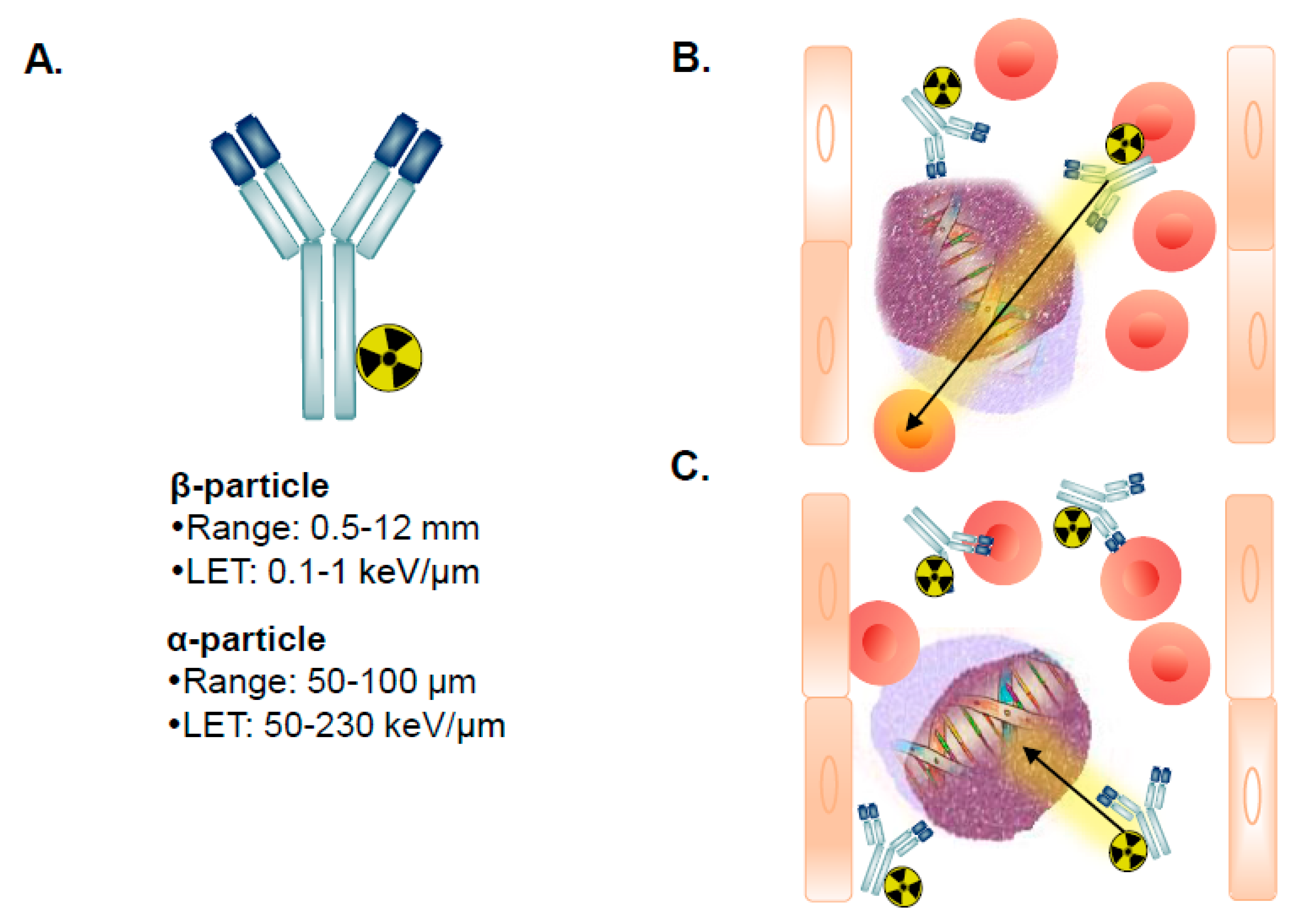
5. Radioimmunotherapy of AML
5.1. β-Particle RIT for AML
5.2. α-Particle RIT for AML
5.3. Ongoing Clinical Trials of RIT in AML
5.4. Future of RIT for AML
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.; Grillo-López, A.J.; Link, B.K.; Levy, R.; Czuczman, M.S.; Williams, M.E.; Heyman, M.R.; Bence-Bruckler, I.; White, C.A.; Cabanillas, F.; et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998, 16, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.W. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother. Rep. 1973, 57, 485–488. [Google Scholar] [PubMed]
- Rai, K.R.; Holland, J.F.; Glidewell, O.J.; Weinberg, V.; Brunner, K.; Obrecht, J.P.; Preisler, H.D.; Nawabi, I.W.; Prager, D.; Carey, R.W.; et al. Treatment of acute myelocytic leukemia: A study by cancer and leukemia group B. Blood 1981, 58, 1203–1212. [Google Scholar] [PubMed]
- Yates, J.; Glidewell, O.; Wiernik, P.; Cooper, M.R.; Steinberg, D.; Dosik, H.; Levy, R.; Hoagland, C.; Henry, P.; Gottlieb, A.; et al. Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: A CALGB study. Blood 1982, 60, 454–462. [Google Scholar] [PubMed]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar]
- Lee, J.H.; Joo, Y.D.; Kim, H.; Bae, S.H.; Kim, M.K.; Zang, D.Y.; Lee, J.L.; Lee, G.W.; Lee, J.H.; Park, J.H.; et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011, 118, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Cavenagh, J.; Kjeldsen, L.; McMullin, M.F.; Cahalin, P.; Dennis, M.; Friis, L.; et al. A randomized comparison of daunorubicin 90 mg/m2 vs. 60 mg/m2 in AML induction: Results from the UK NCRI AML17 trial in 1206 patients. Blood 2015, 125, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin Pharm. 2017, 18, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.D.; Brado, B.; Haas, R.; Hunstein, W. Etoposide in acute leukemia. Past experience and future perspectives. Cancer 1991, 67 (Suppl. 1), 281–284. [Google Scholar] [CrossRef]
- Preisler, H.; Davis, R.B.; Kirshner, J.; Dupre, E.; Hoagland, H.C.; Kopel, S.; Levy, R.N.; Carey, R.; Schulman, P. Comparison of three remission induction regimens and two postinduction strategies for the treatment of acute nonlymphocytic leukemia: A cancer and leukemia group B study. Blood 1987, 69, 1441–1449. [Google Scholar] [PubMed]
- Bishop, J.F.; Lowenthal, R.M.; Joshua, D.; Matthews, J.P.; Todd, D.; Cobcroft, R.; Whiteside, M.G.; Kronenberg, H.; Ma, D.; Dodds, A. Etoposide in acute nonlymphocytic leukemia. Australian Leukemia Study Group. Blood 1990, 75, 27–32. [Google Scholar] [PubMed]
- Clavio, M.; Gatto, S.; Beltrami, G.; Quintino, S.; Canepa, L.; Pierri, I.; Galbusera, V.; Carrara, P.; Miglino, M.; Varaldo, R.; et al. Fludarabine, ARA-C, idarubicin and G-CSF (FLAG-Ida), high dose ARA-C and early stem cell transplant. A feasable and effective therapeutic strategy for de novo AML patients. J. Exp. Clin. Cancer Res. 2002, 21, 481–487. [Google Scholar] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Hunter, A.E.; Kjeldsen, L.; Yin, J.; Gibson, B.E.; Wheatley, K.; Milligan, D. Optimization of chemotherapy for younger patients with acute myeloid leukemia: Results of the medical research council AML15 trial. J. Clin. Oncol. 2013, 31, 3360–3368. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef]
- Lancet, J.E.; Cortes, J.E.; Hogge, D.E.; Tallman, M.S.; Kovacsovics, T.J.; Damon, L.E.; Komrokji, R.; Solomon, S.R.; Kolitz, J.E.; Cooper, M.; et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs. cytarabine/daunorubicin in older adults with untreated AML. Blood 2014, 123, 3239–3246. [Google Scholar] [CrossRef]
- Cortes, J.E.; Goldberg, S.L.; Feldman, E.J.; Rizzeri, D.A.; Hogge, D.E.; Larson, M.; Pigneux, A.; Recher, C.; Schiller, G.; Warzocha, K.; et al. Phase II, multicenter, randomized trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus intensive salvage therapy in adults with first relapse AML. Cancer 2015, 121, 234–242. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassileth, P.A.; Harrington, D.P.; Hines, J.D.; Oken, M.M.; Mazza, J.J.; McGlave, P.; Bennett, J.M.; O’Connell, M.J. Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia. J. Clin. Oncol. 1988, 6, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Davis, R.B.; Schiffer, C.A.; Berg, D.T.; Powell, B.L.; Schulman, P.; Omura, G.A.; Moore, J.O.; McIntyre, O.R.; Frei, E. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N. Engl. J. Med. 1994, 331, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wölfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Rubnitz, J.E. How I treat pediatric acute myeloid leukemia. Blood 2012, 119, 5980–5988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.; Rao, A.V. Current and emerging strategies for the management of acute myeloid leukemia in the elderly. Oncologist 2008, 13, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Khalidi, H.S.; Medeiros, L.J.; Chang, K.L.; Brynes, R.K.; Slovak, M.L.; Arber, D.A. The immunophenotype of adult acute myeloid leukemia: High frequency of lymphoid antigen expression and comparison of immunophenotype, French-American-British classification, and karyotypic abnormalities. Am. J. Clin. Pathol. 1998, 109, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Iriyama, N.; Hatta, Y.; Takeuchi, J.; Ogawa, Y.; Ohtake, S.; Sakura, T.; Mitani, K.; Ishida, F.; Takahashi, M.; Maeda, T.; et al. CD56 expression is an independent prognostic factor for relapse in acute myeloid leukemia with t(8;21). Leuk. Res. 2013, 37, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, E.A.; Till, J.E. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat. Res. 1960, 13, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.L.; Shah, A.J.; Thiemann, F.T.; Smogorzewska, E.M.; Crooks, G.M. A functional comparison of CD34 + CD38- cells in cord blood and bone marrow. Blood 1995, 86, 3745–3753. [Google Scholar] [PubMed]
- Bruce, W.R.; Ash, C.L. Survival of Patients Treated for Cancer of the Breast, Cervix, Lung, and Upper Respiratory Tract at the Ontario Cancer Institute (Toronto) from 1930 to 1957. Radiology 1963, 81, 861–870. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Van Rhenen, A.; Feller, N.; Kelder, A.; Westra, A.H.; Rombouts, E.; Zweegman, S.; Van Der Pol, M.A.; Waisfisz, Q.; Ossenkoppele, G.J.; Schuurhuis, G.J. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin. Cancer Res. 2005, 11, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.E.; Ahlers, J.; Schäfer, I.; André, M.; Kerst, G.; Scheel-Walter, H.G.; Schwarze, C.P.; Pfeiffer, M.; Lang, P.; Handgretinger, R.; et al. High proportion of leukemic stem cells at diagnosis is correlated with unfavorable prognosis in childhood acute myeloid leukemia. Pediatr. Hematol. Oncol. 2011, 28, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34 + CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Jordan, C.T.; LaMere, M.W.; Ashton, J.M.; O’Dwyer, K.; Mendler, J.H.; Liesveld, J.L.; Wang, E.S.; Guzman, M.L.; Calvi, L.M.; et al. A Role for IL1RAP in Acute Myelogenous Leukemia Stem Cells Following Treatment and Progression. Blood 2015, 126, 4266. [Google Scholar]
- Rabinowich, H.; Pricop, L.; Herberman, R.B.; Whiteside, T.L. Expression and function of CD7 molecule on human natural killer cells. J. Immunol. 1994, 152, 517–526. [Google Scholar] [PubMed]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anania, J.C.; Chenoweth, A.M.; Wines, B.D.; Hogarth, P.M. The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease. Front. Immunol. 2019, 10, 464. [Google Scholar] [CrossRef]
- Seddiki, N.; Santner-Nanan, B.; Tangye, S.G.; Alexander, S.I.; Solomon, M.; Lee, S.; Nanan, R.; de Saint Groth, B.F. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood 2006, 107, 2830–2838. [Google Scholar] [CrossRef] [Green Version]
- Pasello, M.; Manara, M.C.; Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal. 2018, 12, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Wang, X.; Jiang, J.; Cheng, X. Involvement of CD244 in regulating CD4+ T cell immunity in patients with active tuberculosis. PLoS ONE 2013, 8, e63261. [Google Scholar] [CrossRef]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From immunoregulatory protein to potential therapeutic target. Immunol. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.A.; Joesting, J.J.; Freund, G.G. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 2013, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, H.; Kane, L.P. Immune regulation by Tim-3. F1000Research 2018, 7, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikushige, Y.; Shima, T.; Takayanagi, S.I.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Lichtenegger, F.S.; Köhnke, T.; Bögeholz, J.; Bücklein, V.; Roiss, M.; Altmann, T.; Do, T.U.; Dusek, R.; Wilson, K.; et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget 2017, 8, 35707–35717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Liu, X.; Chen, C.; Zhu, J.; Yu, Z.; Xie, J.; Xie, L.; Bai, H.; Zhang, Y.; Fang, X.; et al. CD244 maintains the proliferation ability of leukemia initiating cells through SHP-2/p27(kip1) signaling. Haematologica 2017, 102, 707–718. [Google Scholar] [CrossRef]
- Pearce, D.J.; Taussig, D.; Zibara, K.; Smith, L.L.; Ridler, C.M.; Preudhomme, C.; Young, B.D.; Rohatiner, A.Z.; Lister, T.A.; Bonnet, D. AML engraftment in the NOD/SCID assay reflects the outcome of AML: Implications for our understanding of the heterogeneity of AML. Blood 2006, 107, 1166–1173. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; Van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Moretti, S. CD123 (interleukin 3 receptor alpha chain). J. Biol. Regul. Homeost. Agents 2001, 15, 98–100. [Google Scholar]
- Martinez-Moczygemba, M.; Huston, D.P. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J. Allergy Clin. Immunol. 2003, 112, 653–665. [Google Scholar] [PubMed]
- Barry, S.C.; Korpelainen, E.; Sun, Q.; Stomski, F.C.; Moretti, P.A.; Wakao, H.; D’Andrea, R.J.; Vadas, M.A.; Lopez, A.F.; Goodall, G.J. Roles of the N and C terminal domains of the interleukin-3 receptor alpha chain in receptor function. Blood 1997, 89, 842–852. [Google Scholar] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Chen, Z.; Yu, J.F.; Young, D.; Bashey, A.; Ho, A.D.; Law, P. Correlation between IL-3 receptor expression and growth potential of human CD34+ hematopoietic cells from different tissues. Stem Cells 1999, 17, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ashmun, R.A.; Look, A.T. Metalloprotease activity of CD13/aminopeptidase N on the surface of human myeloid cells. Blood 1990, 75, 462–469. [Google Scholar] [PubMed]
- Mina-Osorio, P. The moonlighting enzyme CD13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Piedfer, M.; Dauzonne, D.; Tang, R.; N′Guyen, J.; Billard, C.; Bauvois, B. Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. FASEB J. 2011, 25, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.S.; Willment, J.A.; Lin, H.H.; Williams, D.L.; Gordon, S.; Brown, G.D. Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J. Biol. Chem. 2004, 279, 14792–14802. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar] [PubMed]
- Dianzani, U.; Funaro, A.; DiFranco, D.; Garbarino, G.; Bragardo, M.; Redoglia, V.; Buonfiglio, D.; De Monte, L.B.; Pileri, A.; Malavasi, F. Interaction between endothelium and CD4+CD45RA+ lymphocytes. Role of the human CD38 molecule. J. Immunol. 1994, 153, 952–959. [Google Scholar] [PubMed]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Owens, R.; Tricot, G.; Wilson, C.S. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol. 2004, 121, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Menssen, H.D.; Renkl, H.J.; Rodeck, U.; Maurer, J.; Notter, M.; Schwartz, S.; Reinhardt, R.; Thiel, E. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995, 9, 1060–1067. [Google Scholar] [PubMed]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Ataie, N.; Xiang, J.; Cheng, N.; Brea, E.J.; Lu, W.; Scheinberg, D.A.; Liu, C.; Ng, H.L. Structure of a TCR-Mimic Antibody with Target Predicts Pharmacogenetics. J. Mol. Biol. 2016, 428, 194–205. [Google Scholar] [CrossRef]
- Rheinlander, A.; Schraven, B.; Bommhardt, U. CD45 in human physiology and clinical medicine. Immunol. Lett. 2018, 196, 22–32. [Google Scholar] [CrossRef]
- Hermiston, M.L.; Xu, Z.; Weiss, A. CD45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N. CD45: All is not yet crystal clear. Immunology 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Arancia, G.; Malorni, W.; Donelli, G. Cellular mechanisms of lymphocyte-mediated lysis of tumor cells. Annali Dell’Istituto Superiore di Sanita 1990, 26, 369–384. [Google Scholar] [PubMed]
- McCann, F.E.; Vanherberghen, B.; Eleme, K.; Carlin, L.M.; Newsam, R.J.; Goulding, D.; Davis, D.M. The size of the synaptic cleft and distinct distributions of filamentous actin, ezrin, CD43, and CD45 at activating and inhibitory human NK cell immune synapses. J. Immunol. 2003, 170, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Dennert, G. Assembly of two types of tubules with putative cytolytic function by cloned natural killer cells. Nature 1983, 302, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Dennert, G.; Podack, E.R. Cytolysis by H-2-specific T killer cells. Assembly of tubular complexes on target membranes. J. Exp. Med. 1983, 157, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Zamai, M.; Tschopp, J. Identification of granzyme A isolated from cytotoxic T-lymphocyte-granules as one of the proteases encoded by CTL-specific genes. FEBS Lett. 1986, 208, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Masson, D.; Tschopp, J. A family of serine esterases in lytic granules of cytolytic T lymphocytes. Cell 1987, 49, 679–685. [Google Scholar] [CrossRef]
- Sutton, V.R.; Wowk, M.E.; Cancilla, M.; Trapani, J.A. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity 2003, 18, 319–329. [Google Scholar] [CrossRef]
- Beresford, P.J.; Xia, Z.; Greenberg, A.H.; Lieberman, J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999, 10, 585–594. [Google Scholar] [CrossRef]
- Saxena, R.K.; Saxena, Q.B.; Adler, W.H. Identity of effector cells participating in the reverse antibody-dependent cell-mediated cytotoxicity. Immunology 1982, 46, 459–464. [Google Scholar] [PubMed]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Wang, X.; Routy, B.; Cheng, R.; Maghera, S.; Keating, A. NK Cell Line Killing of Leukemia Cells Is Enhanced By Reverse Antibody Dependent Cell Mediated Cytotoxicity (R-ADCC) Via NKp30 and NKp44 and Target Fcγ Receptor II (CD32). Blood 2014, 124, 2444. [Google Scholar]
- Ball, E.D.; McDermott, J.; Griffin, J.D.; Davey, F.R.; Davis, R.; Bloomfield, C.D. Expression of the three myeloid cell-associated immunoglobulin G Fc receptors defined by murine monoclonal antibodies on normal bone marrow and acute leukemia cells. Blood 1989, 73, 1951–1956. [Google Scholar] [PubMed]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. MAbs 2015, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer 2018, 17, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishelson, Z.; Kirschfink, M. Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 2019, 10, 752. [Google Scholar] [CrossRef] [PubMed]
- He, S.Z.; Busfield, S.; Ritchie, D.S.; Hertzberg, M.S.; Durrant, S.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; Kerridge, I.; Bradstock, K.F.; et al. A Phase 1 study of the Safety, Pharmacokinetics, and Anti-leukemic Activity of the anti-CD123 monoclonal antibody, CSL360, in Relapsed, Refractory or High-Risk Acute Myeloid Leukemia (AML). Leuk. Lymphoma 2014, 56, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Roboz, G.J.; Walter, R.B.; Altman, J.K.; Ferguson, A.; Curcio, T.J.; Orlowski, K.F.; Garrett, L.; Busfield, S.J.; Barnden, M.; et al. First-in Man, Phase 1 Study of CSL362 (Anti-IL3Rα/Anti-CD123 Monoclonal Antibody) in Patients with CD123+ Acute Myeloid Leukemia (AML) in CR at High Risk for Early Relapse. Blood 2014, 124, 120. [Google Scholar]
- Schurch, C.M. Therapeutic Antibodies for Myeloid Neoplasms-Current Developments and Future Directions. Front. Oncol. 2018, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Lancet, J.E.; Wood, B.L.; Grove, L.E.; Sandalic, L.; Sievers, E.L.; Jurcic, J.G. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica 2013, 98, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O’Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Chopra, R.; et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; He, S.; Cheney, C.; Gopalakrishnan, B.; Mani, R.; Lozanski, G.; Mo, X.; Groh, V.; Whitman, S.P.; Konopitzky, R.; et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood 2016, 127, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.C.; Mashayekh, R.; Wymore, K.; McCabe, J.G.; et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994, 368, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Desai, A.; Zeng, D.; Gong, T.; Lu, P.; Wang, M. Magic year for multiple myeloma therapeutics: Key takeaways from the ASH 2015 annual meeting. Oncotarget 2017, 8, 10748–10759. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Xiaochuan, S.; Chenghui, Z.; Ndikuyeze, G.H.; Glover, J.; Secreto, T.; Doshi, P.; Sasser, K.; Danet-Desnoyers, G. Anti-Leukemic Activity of Daratumumab in Acute Myeloid Leukemia Cells and Patient-Derived Xenografts. Blood 2014, 124, 2312. [Google Scholar]
- De Weers, M.; Tai, Y.T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Strickland, S.A.; Glenn, M.; Zheng, W.; Daskalakis, N.; Mikhael, J.R. SAR650984, a CD38 Monoclonal Antibody in Patients with Selected CD38+ Hematological Malignancies- Data From a Dose-Escalation Phase I Study. Blood 2013, 122, 284. [Google Scholar]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67 Pt A, 95–106. [Google Scholar] [CrossRef]
- Handgretinger, R.; Zugmaier, G.; Henze, G.; Kreyenberg, H.; Lang, P.; Von Stackelberg, A. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia 2011, 25, 181–184. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; Stock, W. Taking a “BiTE out of ALL”—Blinatumomab approval for MRD positive ALL. Blood 2019, 16, 1715–1719. [Google Scholar] [CrossRef]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Aigner, M.; Feulner, J.; Schaffer, S.; Kischel, R.; Kufer, P.; Schneider, K.; Henn, A.; Rattel, B.; Friedrich, M.; Baeuerle, P.A.; et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia 2013, 27, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Dell’Aringa, J.; Newhall, K.J.; Means, G.D.; Sinclair, A.M.; Kischel, R.; Frankel, S.R.; Walter, R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014, 123, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol. Cancer 2014, 13, 1549–1557. [Google Scholar] [CrossRef]
- Harrington, K.H.; Gudgeon, C.J.; Laszlo, G.S.; Newhall, K.J.; Sinclair, A.M.; Frankel, S.R.; Kischel, R.; Chen, G.; Walter, R.B. The Broad Anti-AML Activity of the CD33/CD3 BiTE Antibody Construct, AMG 330, Is Impacted by Disease Stage and Risk. PLoS ONE 2015, 10, e0135945. [Google Scholar] [CrossRef]
- Ravandi, F.; Stein, A.S.; Kantarjian, H.M.; Walter, R.B.; Paschka, P.; Jongen-Lavrencic, M.; Ossenkoppele, G.J.; Yang, Z.; Mehta, B.; Subklewe, M. A Phase 1 First-in-Human Study of AMG 330, an Anti-CD33 Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). Blood 2018, 132 (Suppl. 1), 25. [Google Scholar]
- Stamova, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Koristka, S.; Bartsch, H.; Bippes, C.C.; Wehner, R.; Schmitz, M.; von Bonin, M.; et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol. Immunol. 2011, 49, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Von Bonin, M.; Cartellieri, M.; Feldmann, A.; Koristka, S.; Michalk, I.; Stamova, S.; Bornhäuser, M.; Schmitz, M.; Ehninger, G.; et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia 2013, 27, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Feldmann, A.; Von Bonin, M.; Cartellieri, M.; Ewen, E.M.; Koristka, S.; Michalk, I.; Stamova, S.; Berndt, N.; Gocht, A.; et al. Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: Description of a novel modular targeting system. Leukemia 2014, 28, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.R.; Wong, L.; Liu, J.S. Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng. Des. Sel. 2012, 25, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics11Edited by J. Karn. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [Green Version]
- Silla, L.M.; Chen, J.; Zhong, R.K.; Whiteside, T.L.; Ball, E.D. Potentiation of lysis of leukaemia cells by a bispecific antibody to CD33 and CD16 (Fc gamma RIII) expressed by human natural killer (NK) cells. Br. J. Haematol. 1995, 89, 712–718. [Google Scholar] [CrossRef]
- Nitta, T.; Yagita, H.; Azuma, T.; Sato, K.; Okumura KBispecific, F. Bispecific F (ab’)2 monomer prepared with anti-CD3 and anti-tumor monoclonal antibodies is most potent in induction of cytolysis of human T cells. Eur. J. Immunol. 1989, 19, 1437–1441. [Google Scholar] [CrossRef]
- Oshimi, K.; Seto, T.; Oshimi, Y.; Masuda, M.; Okumura, K.O.; Mizoguchi, H. Increased lysis of patient CD10-positive leukemic cells by T cells coated with anti-CD3 Fab’ antibody cross-linked to anti-CD10 Fab’ antibody. Blood 1991, 77, 1044–1049. [Google Scholar]
- Kaneko, T.; Fusauchi, Y.; Kakui, Y.; Masuda, M.; Akahoshi, M.; Teramura, M.; Motoji, T.; Okumura, K.; Mizoguchi, H.; Oshimi, K. A bispecific antibody enhances cytokine-induced killer-mediated cytolysis of autologous acute myeloid leukemia cells. Blood 1993, 81, 1333–1341. [Google Scholar] [PubMed]
- Gaudet, F.; Nemeth, J.F.; McDaid, R.; Li, Y.; Harman, B.; Millar, H.; Teplyakov, A.; Wheeler, J.; Luo, J.; Tam, S.; et al. Development of a CD123xCD3 Bispecific Antibody (JNJ-63709178) for the Treatment of Acute Myeloid Leukemia (AML). Blood 2016, 128, 2824. [Google Scholar]
- Van Loo, P.F.; Doornbos, R.; Dolstra, H.; Shamsili, S.; Bakker, L. Preclinical Evaluation of MCLA117, a CLEC12AxCD3 Bispecific Antibody Efficiently Targeting a Novel Leukemic Stem Cell Associated Antigen in AML. Blood 2015, 126, 325. [Google Scholar]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Singer, H.; Kellner, C.; Lanig, H.; Aigner, M.; Stockmeyer, B.; Oduncu, F.; Schwemmlein, M.; Stein, C.; Mentz, K.; Mackensen, A.; et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J. Immunother 2010, 33, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kügler, M.; Stein, C.; Kellner, C.; Mentz, K.; Saul, D.; Schwenkert, M.; Schubert, I.; Singer, H.; Oduncu, F.; Stockmeyer, B.; et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br. J. Haematol. 2010, 150, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Ghose, T.; Norvell, S.T.; Guclu, A.; MacDonald, A.S. Immunochemotherapy of human malignant melanoma with chlorambucil-carrying antibody. Eur J. Cancer 1975, 11, 321–326. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, W.D.; Fandy, T.E.; Ashour, H.M.; Wang, H.; Hassan, H.E. Antibody-Drug Conjugates: Pharmacokinetic/Pharmacodynamic Modeling, Preclinical Characterization, Clinical Studies, and Lessons Learned. Clin. Pharm. 2018, 57, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201. [Google Scholar] [CrossRef]
- Richardson, N.C.; Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Brentuximab Vedotin in First-Line Treatment of Peripheral T-Cell Lymphoma. Oncologist 2019, 24, e180–e187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregson, S.J.; Masterson, L.A.; Wei, B.; Pillow, T.H.; Spencer, S.D.; Kang, G.D.; Yu, S.F.; Raab, H.; Lau, J.; Li, G.; et al. Pyrrolobenzodiazepine Dimer Antibody-Drug Conjugates: Synthesis and Evaluation of Noncleavable Drug-Linkers. J. Med. Chem 2017, 60, 9490–9507. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Harrington, K.H.; Stone, I.; Ryan, M.C.; Sussman, D.; Zeng, W.; et al. SGN-CD33A: A Novel CD33-Directed Antibody-Drug Conjugate, Utilizing Pyrrolobenzodiazepine Dimers, Demonstrates Preclinical Antitumor Activity Against Multi-Drug Resistant Human AML. Blood 2012, 120, 3589. [Google Scholar]
- Smellie, M.; Bose, D.S.; Thompson, A.S.; Jenkins, T.C.; Hartley, J.A.; Thurston, D.E. Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: Kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers. Biochemistry 2003, 42, 8232–8239. [Google Scholar] [CrossRef]
- van der Velden, V.H.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Bixby, D.L.; Stein, A.S.; Fathi, A.T.; Kovacsovics, T.J.; Levy, M.Y.; Erba, H.P.; Lancet, J.E.; Jillella, A.P.; Ravandi, F.; Walter, R.B.; et al. Vadastuximab Talirine Monotherapy in Older Patients with Treatment Naive CD33-Positive Acute Myeloid Leukemia (AML). Blood 2016, 128, 590. [Google Scholar]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.; DeAngelo, D.J.; Kovacsovics, T.J.; et al. Vadastuximab Talirine Plus Hypomethylating Agents: A Well-Tolerated Regimen with High Remission Rate in Frontline Older Patients with Acute Myeloid Leukemia (AML). Blood 2016, 128, 591. [Google Scholar]
- Whiteman, K.R.; Noordhuis, P.; Walker, R.; Watkins, K.; Kovtun, Y.; Harvey, L.; Wilhelm, A.; Johnson, H.; Schuurhuis, G.J.; Ossenkoppele, G.J.; et al. The Antibody-Drug Conjugate (ADC) IMGN779 Is Highly Active in Vitro and in Vivo Against Acute Myeloid Leukemia (AML) with FLT3-ITD Mutations. Blood 2014, 124, 2321. [Google Scholar]
- Wu, A.M.; Senter, P.D. Arming antibodies: Prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour. Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.P.; Xie, C.; Ding, N.; Zhang, Y.J.; Han, L.; Han, Y.W. Targeted inhibition of genome-wide DNA methylation analysis in epigenetically modulated phenotypes in lung cancer. Med. Oncol. 2015, 32, 615. [Google Scholar] [CrossRef]
- Aghevlian, S.; Boyle, A.J.; Reilly, R.M. Radioimmunotherapy of cancer with high linear energy transfer (LET) radiation delivered by radionuclides emitting alpha-particles or Auger electrons. Adv. Drug Deliv. Rev. 2017, 109, 102–118. [Google Scholar] [CrossRef]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.K.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Rizvi, S.M.; Henniker, A.J.; Goozee, G.; Allen, B.J. In vitro testing of the leukaemia monoclonal antibody WM-53 labeled with alpha and beta emitting radioisotopes. Leuk. Res. 2002, 26, 37–43. [Google Scholar] [CrossRef]
- Heylmann, D.; Rödel, F.; Kindler, T.; Kaina, B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim. Biophys. Acta 2014, 1846, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kersemans, V.; Cornelissen, B.; Minden, M.D.; Brandwein, J.; Reilly, R.M. Drug-resistant AML cells and primary AML specimens are killed by 111In-anti-CD33 monoclonal antibodies modified with nuclear localizing peptide sequences. J. Nucl. Med. 2008, 49, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.V.; Gao, C.; Williams, B.; Keating, A.; Minden, M.; Reilly, R.M. A radiolabeled antibody targeting CD123(+) leukemia stem cells—initial radioimmunotherapy studies in NOD/SCID mice engrafted with primary human AML. Leuk. Res. Rep. 2015, 4, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.V.; Hu, M.; Gao, C.; Turner, P.V.; Dick, J.E.; Minden, M.; Reilly, R.M. Auger electron radioimmunotherapeutic agent specific for the CD123+/CD131- phenotype of the leukemia stem cell population. J. Nucl. Med. 2011, 52, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.V.; Williams, B.; Gao, C.; Keating, A.; Minden, M.; Reilly, R.M. MicroSPECT/CT imaging of primary human AML engrafted into the bone marrow and spleen of NOD/SCID mice using 111In-DTPA-NLS-CSL360 radioimmunoconjugates recognizing the CD123+/CD131- epitope expressed by leukemia stem cells. Leuk. Res. 2014, 38, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Gyurkocza, B.; Sandmaier, B.M. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 2014, 124, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Wallen, H.; Gooley, T.A.; Deeg, H.J.; Pagel, J.M.; Press, O.W.; Appelbaum, F.R.; Storb, R.; Gopal, A.K. Ablative allogeneic hematopoietic cell transplantation in adults 60 years of age and older. J. Clin. Oncol. 2005, 23, 3439–3446. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, P.A.; Niederwieser, D.; Shizuru, J.A.; Sandmaier, B.M.; Molina, A.J.; Maloney, D.G.; Chauncey, T.R.; Gooley, T.A.; Hegenbart, U.; Nash, R.A.; et al. Hematopoietic cell transplantation in older patients with hematologic malignancies: Replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood 2001, 97, 3390–3400. [Google Scholar] [CrossRef]
- Clift, R.A.; Buckner, C.D.; Appelbaum, F.R.; Bearman, S.I.; Petersen, F.B.; Fisher, L.D.; Anasetti, C.; Beatty, P.; Bensinger, W.I.; Doney, K. Allogeneic marrow transplantation in patients with acute myeloid leukemia in first remission: A randomized trial of two irradiation regimens. Blood 1990, 76, 1867–1871. [Google Scholar]
- Clift, R.A.; Buckner, C.D.; Appelbaum, F.R.; Sullivan, K.M.; Storb, R.; Thomas, E.D. Long-term follow-Up of a randomized trial of two irradiation regimens for patients receiving allogeneic marrow transplants during first remission of acute myeloid leukemia. Blood 1998, 92, 1455–1456. [Google Scholar] [PubMed]
- Goyal, G. Reduced-intensity conditioning allogeneic hematopoietic-cell transplantation for older patients with acute myeloid leukemia. Adv. Hematol. 2016, 7, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storb, R.; Gyurkocza, B.; Storer, B.E.; Sorror, M.L.; Blume, K.; Niederwieser, D.; Chauncey, T.R.; Pulsipher, M.A.; Petersen, F.B.; Sahebi, F.; et al. Graft-versus-host disease and graft-versus-tumor effects after allogeneic hematopoietic cell transplantation. J. Clin. Oncol. 2013, 31, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, F.; Durrieu, F.; Briais, A.; Dumain, P.; Belloc, F.; Bascans, E.; Reiffers, J.; Boisseau, M.R.; Bernard, P. Flow cytometry CD45 gating for immunophenotyping of acute myeloid leukemia. Leukemia 1997, 11, 1878–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, D.C.; Appelbaum, F.R.; Eary, J.F.; Fisher, D.R.; Durack, L.D.; Hui, T.E.; Martin, P.J.; Mitchell, D.; Press, O.W.; Storb, R.; et al. Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood 1999, 94, 1237–1247. [Google Scholar] [PubMed]
- Pagel, J.M.; Appelbaum, F.R.; Eary, J.F.; Rajendran, J.; Fisher, D.R.; Gooley, T.; Ruffner, K.; Nemecek, E.; Sickle, E.; Durack, L.; et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood 2006, 107, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.M.; Gooley, T.A.; Rajendran, J.; Fisher, D.R.; Wilson, W.A.; Sandmaier, B.M.; Matthews, D.C.; Deeg, H.J.; Gopal, A.K.; Martin, P.J.; et al. Allogeneic hematopoietic cell transplantation after conditioning with 131I-anti-CD45 antibody plus fludarabine and low-dose total body irradiation for elderly patients with advanced acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood 2009, 114, 5444–5453. [Google Scholar] [CrossRef]
- Mawad, R.; Gooley, T.A.; Rajendran, J.G.; Fisher, D.R.; Gopal, A.K.; Shields, A.T.; Sandmaier, B.M.; Sorror, M.L.; Deeg, H.J.; Storb, R.; et al. Radiolabeled anti-CD45 antibody with reduced-intensity conditioning and allogeneic transplantation for younger patients with advanced acute myeloid leukemia or myelodysplastic syndrome. Biol. Blood Marrow Transpl. 2014, 20, 1363–1368. [Google Scholar] [CrossRef]
- Orozco, J.J.; Kenoyer, A.; Balkin, E.R.; Gooley, T.A.; Hamlin, D.K.; Wilbur, D.S.; Hylarides, M.D.; Frost, S.H.; Mawad, R.; O’Donnell, P.; et al. Anti-CD45 radioimmunotherapy without TBI before transplantation facilitates persistent haploidentical donor engraftment. Blood 2016, 127, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Orozco, J.; Zeller, J.; Pagel, J.M. Radiolabeled antibodies directed at CD45 for conditioning prior to allogeneic transplantation in acute myeloid leukemia and myelodysplastic syndrome. Adv. Hematol. 2011, 3, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Agura, E.D.; Gyurkocza, B.; Nath, R.; Litzow, M.R.; Tomlinson, B.K.; Abhyankar, S.; Seropian, S.; Stiff, P.J.; Choe, H.K.; Kebriaei, P.; et al. Targeted Conditioning of Iomab-B (131I-anti-CD45) Prior to Allogeneic Hematopoietic Cell Transplantation Versus Conventional Care in Relapsed or Refractory Acute Myeloid Leukemia (AML): Preliminary Feasibility and Safety Results from the Prospective, Randomized Phase 3 Sierra Trial. Blood 2018, 132 (Suppl. 1), 1017. [Google Scholar]
- Cassaday, R.D.; Press, O.W.; Pagel, J.M.; Rajendran, J.G.; Gooley, T.A.; Fisher, D.R.; Miyaoka, R.S.; Maloney, D.G.; Smith, S.D.; Till, B.G.; et al. Safety and Efficacy of Escalating Doses of 90Y-BC8-DOTA (Anti-CD45) Followed by Carmustine, Etoposide, Cytarabine, and Melphalan (BEAM) Chemotherapy and Autologous Stem Cell Transplantation (ASCT) for High- Risk Lymphoma. Biol. Blood Marrow Transpl. 2018, 24, 329. [Google Scholar] [CrossRef]
- Glatting, G.; Müller, M.; Koop, B.; Hohl, K.; Friesen, C.; Neumaier, B.; Berrie, E.; Bird, P.; Hale, G.; Blumstein, N.M.; et al. Anti-CD45 monoclonal antibody YAML568: A promising radioimmunoconjugate for targeted therapy of acute leukemia. J. Nucl. Med. 2006, 47, 1335–1341. [Google Scholar] [PubMed]
- Jurcic, J.G.; Larson, S.M.; Sgouros, G.; McDevitt, M.R.; Finn, R.D.; Divgi, C.R.; Ballangrud, Å.M.; Hamacher, K.A.; Ma, D.; Humm, J.L.; et al. Targeted alpha particle immunotherapy for myeloid leukemia. Blood 2002, 100, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.M.; Finn, R.D.; Larson, S.M.; Scheinberg, D.A. Pharmacokinetics and dosimetry of an alpha-particle emitter labeled antibody: 213Bi-HuM195 (anti-CD33) in patients with leukemia. J. Nucl. Med. 1999, 40, 1935–1946. [Google Scholar]
- Jurcic, J.G.; Rosenblat, T.L.; McDevitt, M.R.; Pandit-Taskar, N.; Carrasquillo, J.A.; Chanel, S.M.; Ryan, C.; Frattini, M.G.; Cicic, D.; Larson, S.M.; et al. Phase I Trial of the Targeted Alpha-Particle Nano-Generator Actinium-225 (225Ac)-Lintuzumab (Anti-CD33; HuM195) in Acute Myeloid Leukemia (AML). Blood 2011, 118, 6516. [Google Scholar] [CrossRef]
- Jurcic, J.G.; Ravandi, F.; Pagel, J.M.; Park, J.H.; Smith, B.D.; Douer, D.; Levy, M.Y.; Estey, E.; Kantarjian, H.M.; Earle, D.; et al. Phase I trial of a-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia. J. Clin. Oncol. 2017, 33, 7050. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Rausch, C.R.; Benton, C.; Kadia, T.; Jain, N.; Pemmaraju, N.; Daver, N.; Covert, W.; Marx, K.R.; Mace, M.; et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 2018, 93, 401–407. [Google Scholar] [CrossRef]
- Wierzbowska, A.; Robak, T.; Pluta, A.; Wawrzyniak, E.; Cebula, B.; Hołowiecki, J.; Kyrcz-Krzemień, S.; Grosicki, S.; Giebel, S.; Skotnicki, A.B.; et al. Cladribine combined with high doses of arabinoside cytosine, mitoxantrone, and G-CSF (CLAG-M) is a highly effective salvage regimen in patients with refractory and relapsed acute myeloid leukemia of the poor risk: A final report of the Polish Adult Leukemia Group. Eur. J. Haematol. 2008, 80, 115–126. [Google Scholar]
- Muppidi, M.R.; Freyer, C.W.; Ford, L.A.; Ontiveros, E.P.; Thompson, J.E.; Griffiths, E.A.; Wang, E.S. CLAG±M (cladribine, cytarabine, granulocyte colony stimulating factor ± mitoxantrone) Results in High Response Rates in Older Patients with Secondary and Relapsed/Refractory Acute Myeloid Leukemia—A Single Institute Experience. Blood 2015, 126, 1341. [Google Scholar]
- Li, T.; Ao, E.C.; Lambert, B.; Brans, B.; Vandenberghe, S.; Mok, G.S. Quantitative Imaging for Targeted Radionuclide Therapy Dosimetry—Technical Review. Theranostics 2017, 7, 4551–4565. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Capala, J.; Ivy, S.P. Radiopharmaceuticals for Relapsed or Refractory Leukemias. Front. Oncol. 2019, 9, 97. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NCI Clinical Trial and Phase | Target | Agent(s) | Inclusion Criteria | Estimated Start and End Dates | Outcomes | Status |
|---|---|---|---|---|---|---|
| NCT02715011 Phase 1 | CD123 | JNJ-63709178 CD123 × CD3 Duobody | ≥18 years of age with R/R AML | June 2016 October 2021 | MTD, ORR, 1.5 year EFS and RFS | Suspended |
| NCT02520427 Phase 1 | CD33 | AMG330 CD33 × CD3 Tandem scFv (BiTE) | ≥18 years of age with R/R AML | August 2015 January 2020 | MTD and ORR duration at 3 years | Suspended |
| NCT02152956 Phase 1 | CD123 | MGD006 CD123 × CD3 DART flotetuzumab | ≥18 years of age with R/R AML | May 2014 April 2020 | MTD and OS at 2 years | Recruiting |
| NCI Clinical Trial and Phase | Target | Agent(s) | Inclusion Criteria | Estimated Start and End Dates | Status |
|---|---|---|---|---|---|
| NCT03374332 Phase 2 | CD33 | gemtuzmab ozogamicin | ≥18 years of age with R/R AML | June 2019 March 2021 | Not yet recruiting |
| NCT03737955 Phase 2 | CD33 | gemtuzmab ozogamicin | ≥2 years of age with AML in CR with MRD after induction chemotherapy | November 2018 August 2021 | Recruiting |
| NCT03531918 Phase 1/2 | CD33 | gemtuzmab ozogamicin in combination with GCLAM | ≥18 years of age with untreated “high-grade” myeloid neoplasm (≥10% Blasts in blood or BM) or AML, exluding APL | September 2018 July 2025 | Recruiting |
| NCT02674763 Phase 1 | CD33 | IMGN779 | ≥18 years of age with R/R AML | March 2016 December 2019 | Recruiting |
| NCT03386513 Phase 1 | CD123 | IMGN632 | ≥18 years of age with R/R CD123 + AML and other CD123 + malignancies | January 2018 February 2021 | Recruiting |
| NCT02864290 Phase 1 | FLT3 | ASP1235 (AGS62P1) | ≥18 years of age with R/R AML | November 2016 January 2024 | Recruiting |
| NCT03957915 Phase 1 | CD71 | INA03 | ≥18 years of age R/R AML, ALL, or MPAL with ≥ 20% CD71 positive blasts | September 2019 November 2021 | Active, not recruiting |
| NCT01830777 Phase 1 | CD30 | Brentuximab vedotin in combination with Mitoxantrone, Etoposide, and Cytarabine | ≥18 years of age with CD30 + relapsed AML | May 2013 December 2019 | Active, not recruiting |
| Radionuclide | T ½ | Emission | Emax (keV) | Range (µm) |
|---|---|---|---|---|
| β-Emitting Radionuclides (LET = 0.1–1.0 keV/µm) | ||||
| Iodine-131 | 8.02 days | β and γ | 610/362 | 2300 |
| Yttrium-90 | 2.67 days | β | 2250 | 11,300 |
| α-Emitting Radionuclides (LET = 50–230 keV/µm) | ||||
| Astatine-211 | 7.2 h | α and X | 5870 and 7450/ 77–92 | 80 |
| Actinium-225 | 9.92 days | 4α, 2β and γ | 6000–8000/ 198–659/218–444 | 90 |
| Bismuth-213 | 45.59 min | α and γ | 8400/440 | 17 |
| NCI Clinical Trial and Phase | Target | Agent(s) | Inclusion Criteria | Estimated Start and End Dates | Outcomes | Status |
|---|---|---|---|---|---|---|
|
NCT02665065
(SIERRA) Phase 3 | CD45 | 131I-BC8 Fludarabine 2-Gy TBI | ≥55 years of age with R/R AML patients | June 2015 June 2020 | Durable CR and OS at 1 year | Recruiting |
| NCT03867682 Phase 1/2 | CD33 | 225Ac-lintuzumab Venetoclax Spironolactone | ≥18 years of age with refractory R/R AML. | May 2019 November 2022 | MTD and complete and partial remission status at 6, 12, and 24 months | Not yet recruiting |
| NCT03670966 Phase 1/2 | CD45 | 211At-BC8 Fludarabine Cyclophosphamide 2-Gy TBI Haplotype transplant | ≥18 years of age with R/RAML who have an available haploindentical donor for a haplo HSCT. | March 2019 September 2024 | Toxicity (GVHD, and NRM), donor chimerism, rate of engraftment, and OS up to 100 days and maintenance of remission at 2 years | Recruiting |
| NCT03128034 Phase 1/2 | CD45 | 211At-BC8 Fludarabine 2-3-Gy TBI Haplotype transplant | ≥18 years of age with R/R AML who have an available haploindentical donor for a haplo HSCT. | October 2017 March 2023 | Toxicity (GVHD, and NRM), donor chimerism, rate of engraftment, and OS up to 100, remission at 2 years | Recruiting |
| NCT03441048 Phase 1 | CD45 | 211At-BC8 CLAG-M (cladribine, cytarabine, G-CSF, mitoxantrone) | ≥18 years of age with R/R AML | May 2018 October 2020 | MTD and toxicity | Recruiting |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, B.A.; Law, A.; Hunyadkurti, J.; Desilets, S.; Leyton, J.V.; Keating, A. Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats. J. Clin. Med. 2019, 8, 1261. https://doi.org/10.3390/jcm8081261
Williams BA, Law A, Hunyadkurti J, Desilets S, Leyton JV, Keating A. Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats. Journal of Clinical Medicine. 2019; 8(8):1261. https://doi.org/10.3390/jcm8081261
Chicago/Turabian StyleWilliams, Brent A., Arjun Law, Judit Hunyadkurti, Stephanie Desilets, Jeffrey V. Leyton, and Armand Keating. 2019. "Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats" Journal of Clinical Medicine 8, no. 8: 1261. https://doi.org/10.3390/jcm8081261