Accumulation of prion protein in tonsil and appendix: review of tissue samples
BMJ 2002; 325 doi: https://doi.org/10.1136/bmj.325.7365.633 (Published 21 September 2002) Cite this as: BMJ 2002;325:633
- David A Hilton (david.hilton{at}phnt.swest.nhs.uk), neuropathologista,
- Azra C Ghani, research fellowb,
- Lisa Conyers, research techniciana,
- Philip Edwards, biomedical scientista,
- Linda McCardle, biomedical scientistc,
- Mark Penney, research techniciana,
- Diane Ritchie, research assistantc,
- James W Ironside, professor of clinical neuropathologyc
- a Department of Histopathology, Derriford Hospital, Plymouth PL6 8DH
- b Department of Infectious Disease Epidemiology, Faculty of Medicine, Imperial College of Science, Technology and Medicine, London SW7 2AZ
- c National CJD Surveillance Unit, University of Edinburgh, Edinburgh EH4 2XU
- Correspondence to: D A Hilton
- Accepted 30 July 2002
Variant Creutzfeldt-Jakob disease is almost certainly caused by the bovine spongiform encephalopathy agent, and although the disease is rare (115 deaths to date) there is uncertainty about future numbers of cases.1 The lack of a conventional immune response and the inability to detect abnormal prion protein in blood has hampered the development of a blood test.1 Lymphoreticular accumulation of prion protein has been used as a preclinical test for scrapie (the form of the disease in sheep and goats) and is a consistent feature of variant Creutzfeldt-Jakob disease, occurring before the onset of symptoms.2–4 We screened large numbers of specimens from appendicectomies and tonsillectomies for the presence of prion protein in lymphoreticular tissue to determine the number of people with preclinical variant Creutzfeldt-Jakob disease.
Methods and results
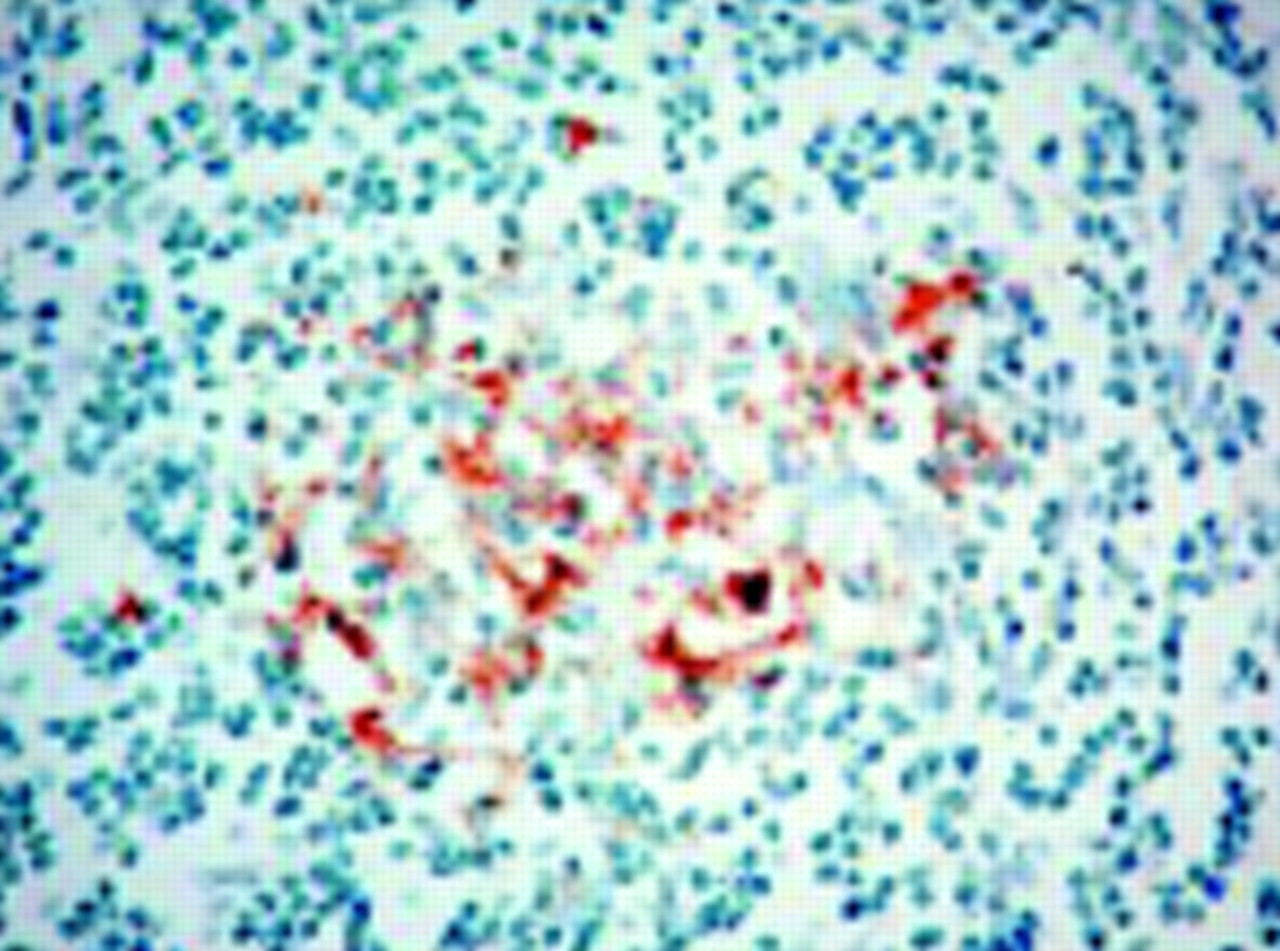
We retrieved appendix and tonsil samples removed between 1995 and 1999 from patients aged 10-50 from histopathology departments across the United Kingdom. The samples were anonymised before testing. We also examined appendix samples removed at autopsy or at surgery from patients with variant Creutzfeldt-Jakob disease. Prion protein was detected by immunohistochemistry with monoclonal antibodies 3F4 (Dako, Ely, Cambridge) and KG9 (Institute for Animal Health, Newbury) and visualised with the catalysed signal amplification system (Dako). We excluded cases with fewer than five secondary lymphoid follicles from the final analyses because in the previously reported case and those examined at autopsy, prion protein was present in around 20% of follicles.4 We tested 11 228 samples, of which 2910 were excluded. Most samples were appendixes and 70% were from patients aged 20-29. In one case we identified prion protein immunoreactivity in a single lymphoid follicle stained with KG9 (figure). The pattern of staining was similar to that seen in the two other patients who developed variant Creutfeldt-Jakob disease (figure1).4 This positive staining was less evident in sections stained with 3F4 antibody. The reason for this discrepancy is not clear but may be due to sampling error because of the focal nature of prion protein deposition. In the appendixes removed before the onset of symptoms in three patients with variant Creutfeldt-Jakob disease, two (removed in 1995 and 1996; see figure) were positive, and the third (removed in 1987) was negative.4 Overall, 19 of 20 appendixes removed at autopsy from cases of variant Creutzfeldt-Jakob disease with adequate numbers of lymphoid follicles had lymphoreticular accumulation of prion protein.

(Left) Distribution of coarse granular prion protein within lymphoid follicle of appendix, suggestive of follicular dendritic cells. (Right) Lymphoreticular accumulation of prion protein in appendix tissue from patient with Creutzfeldt-Jakob disease, removed two years before onset of symptoms
{kind=link}

(Left) Distribution of coarse granular prion protein within lymphoid follicle of appendix, suggestive of follicular dendritic cells. (Right) Lymphoreticular accumulation of prion protein in appendix tissue from patient with Creutzfeldt-Jakob disease, removed two years before onset of symptoms
{kind=link}
Comment
One appendix showing the lymphoreticular accumulation of prion protein out of 8318 samples tested gives an estimated detectable prevalence of prion protein accumulation of 120 per million (95% confidence interval, 0.5 to 900) among people aged 10-50 between 1995 and 1999. Our study design precluded transmission studies, but the accumulation of prion protein in lymphoreticular tissue detected by the immunohistochemical technique correlates with the detection of abnormal prion protein by western blotting and remains the only technique shown to reliably predict disease in animals. 2 3 5 The sensitivity and specificity of preclinical lymphoreticular accumulation of prion protein in predicting variant Creutzfeldt-Jakob disease are unknown, but in sheep exposed to scrapie are estimated to be 87% and 94%, respectively.5 At autopsy, 95% of appendixes from patients with variant Creutzfeldt-Jakob disease with adequate amounts of lymphoid tissue showed accumulation of prion protein, as did both appendixes removed during the 1990s from patients who later developed the disease.
We provide the first estimate of the number of people who may be a potential source of variant Creutzfeldt-Jakob disease by iatrogenic spread. Large scale prospective screening of tissue from tonsillectomies is needed to obtain precise data on prevalence. The use of fresh tissue would allow for a semi-automated western blot assay and verification of positive samples by transmission studies. As half of tonsillectomies are in children under 10, who will have had little or no exposure to bovine spongiform encephalopathy, the opportunity for such a study will diminish over time.
Acknowledgments
We thank histopathology departments and relatives of patients with variant Creutzfeldt-Jakob disease who gave consent for autopsy tissues to be used as positive control material.
Contributors: DAH designed the English study, screened samples tested at Plymouth, and drafted the manuscript; he will act as guarantor for the paper. JWI designed the Scottish study, screened samples tested at Edinburgh, and contributed to the manuscript. ACG performed the statistical analyses and contributed to the manuscript. MP, LC, and PE performed the technical work at Plymouth. LMcC and DR performed the technical work and assisted in screening samples at Edinburgh.
Footnotes
-
Funding Department of Health and the Royal Society. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
-
Competing interests None declared.