Article Text

Statistics from Altmetric.com
At a population level, several environmental, lifestyle and genetic risk factors contribute to the development of coronary artery disease (CAD) and heart failure. However, at an individual level, both susceptibility to, and the age of onset of, these conditions vary considerably even for subjects with apparently similar risk factor profiles. Any mechanism that is proposed to explain this interindividual variation needs to take into account the age association of these diseases (ie, that they are more common with age) and integrate the impact of known risk factors. Here we discuss emerging evidence that suggests that at least part of the interindividual difference in susceptibility to, and in age of onset of, CAD, and possibly other cardiovascular diseases including heart failure, reflects interindividual variation in biological ageing and that mean telomere length acts as a valuable marker of this process.
TELOMERES
Telomeres are the extreme ends of eukaryotic chromosomes. They are made up of a large number of tandem repeats of a simple DNA sequence (in humans TTAGGG). Telomeres are important structures involved in cell cycle control and maintenance of chromosomal stability.1 2 They have three features that mark them as possible markers of interindividual variation in biological ageing:
-
The numbers of repeats (and hence telomere lengths) have a significant genetic determination and vary between individual subjects at birth and through life.3–6
-
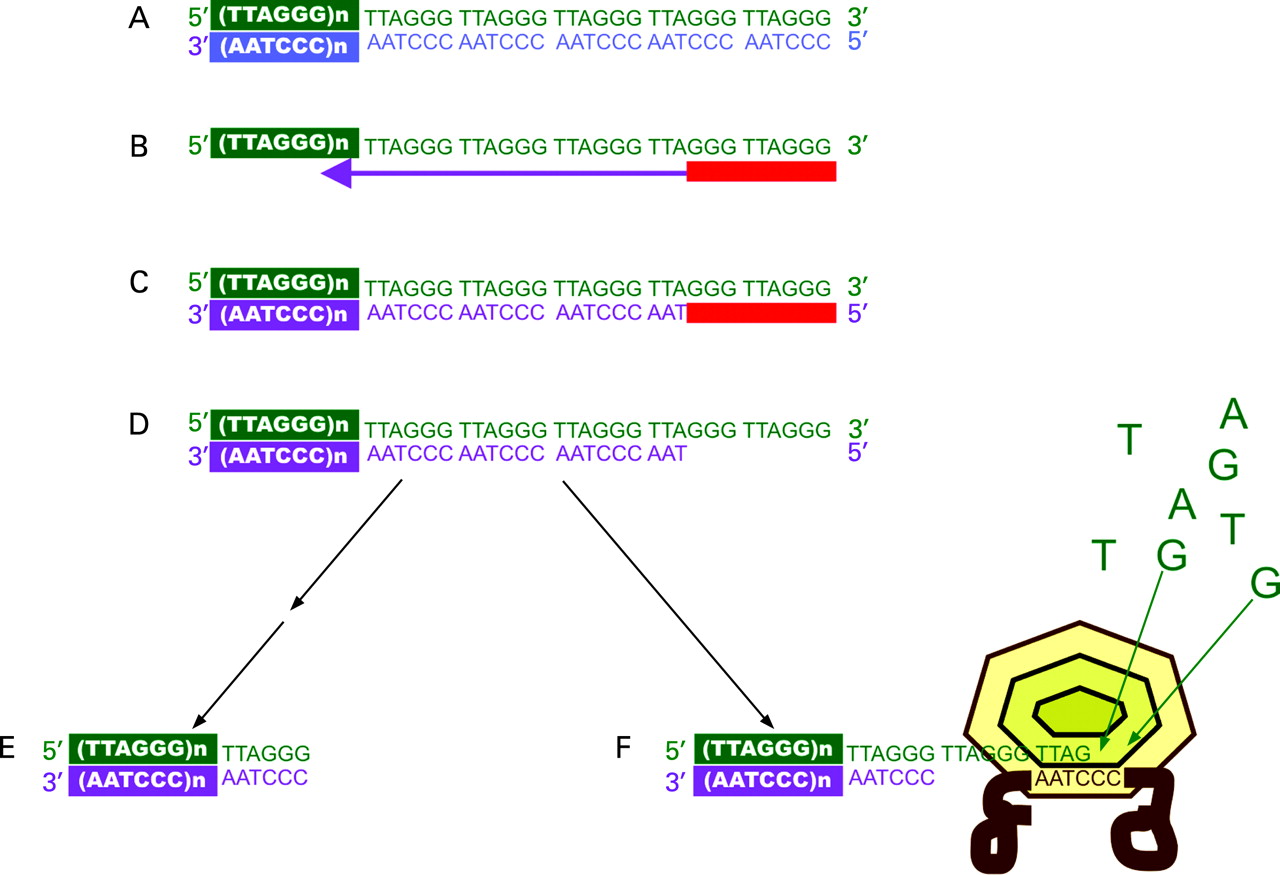
In somatic cells telomeres shorten progressively with repeated cell division.7 This is because DNA polymerase cannot fully replicate the 3′ end of linear DNA during mitosis, the so-called end replication problem (fig 1), and some telomeric DNA is lost during each division.1 2 In utero, in stems cells, and in many cancer cells, the enzyme telomerase helps to maintain the telomere (fig 1) but telomerase is transcriptionally repressed in most somatic cells.1 2
-
The amount of telomere lost for each cell division can vary. In particular, there is evidence that increased oxidative stress, which is a feature of many cardiovascular diseases, is associated with greater telomere loss.8–10

These features provide a potential mechanism for cellular behaviour determined by a biological clock and this concept is supported by strong experimental evidence.11–13 In many cell types senescence and subsequent cell death often occurs when the mean telomere length reaches a critical value.12 More recent evidence suggests that transition to the senescent state may be primarily driven by signals coming from the most shortened telomeres.1 2 Nonetheless, mean telomere length has emerged as a possible marker for biological age, at least at the cellular level, with shorter telomeres indicating increased biological age.13
ASSOCIATION OF SHORTER TELOMERES WITH CORONARY ARTERY DISEASE
Several studies in diverse populations have now shown an association between shorter telomeres in circulating leucocytes and CAD.14–18 Particularly persuasive in this regard are studies that have shown the presence of shorter telomeres before the development of clinical disease, indicating that the shorter telomeres in such subjects are not simply a consequence of CAD. In the West of Scotland Primary Prevention Study (WOSCOPS), the odds ratio for coronary events over the next 4.9 years was almost doubled in placebo-treated subjects in the lower two tertiles of baseline telomere length compared with the highest,18 while a study of 143 people over the age of 60 found that having shorter than average telomeres was associated with a more than threefold higher cardiac mortality over the next 8.9 years.16 Where examined, the difference in telomere lengths in subjects with and without CAD is not explained by differences in classical risk factors for CAD.15 17 18
Because telomeres shorten with age and the average amount of telomeres lost per year can be computed, another way of presenting the difference in telomere length in those with (or who will develop) CAD and those without CAD is by the number of years of age to which this equates. Studies to date, except in very old subjects, have shown a consistent difference equivalent to between 8 and 12 years (ie, on average those with CAD have telomeres equivalent in size to normal subjects 8–12 years older).14 15 18 This remarkable observation provides persuasive evidence that subjects with (or prone to) CAD may be “biologically” older.
TELOMERE LENGTH IN OTHER CARDIOVASCULAR DISEASES
Fewer studies have investigated the association of telomere length with other cardiovascular diseases. However, in the Cardiovascular Health Study, an association with stroke of a similar magnitude to that with myocardial infarction was observed, although there was no association with development of symptomatic peripheral vascular disease.17 Shorter leucocyte mean telomere length has also been reported to be associated with an increased predilection to carotid artery atherosclerosis in hypertensive subjects.19 Recent studies also suggest a role for telomeres in the pathophysiology of congestive heart failure (CHF). In a large cohort of 620 patients with CHF derived from the MERIT-HF study and 183 age- and gender-balanced controls, telomere length of white blood cells was also about 40% reduced in patients with CHF and related to the severity of disease.20 Decreased left ventricular ejection fraction in the elderly, without evidence of previous myocardial infarction, has also been associated with reduced telomere length. One standard deviation of shorter telomeres was associated with a 5% lower ejection fraction.21
TELOMERE LENGTH AND CARDIOVASCULAR RISK FACTORS
Apart from age, several other demographic and conventional risk factors also show an association with telomere length in circulating white cells. Age-adjusted telomere lengths are longer in women than in men,10 22 which may reflect the effect of oestrogens on telomerase.23 In men, telomeres have been shown to be shorter in subjects with type 1 diabetes,24 type 2 diabetes25 and insulin resistance.9 Telomere length has been reported to be inversely correlated with pulse pressure and pulse wave velocity, especially in men.22 In women psychological stress,26 obesity and smoking27 and low socioeconomic status28 have been associated with shorter leucocyte telomeres. Many of these associations could reflect the effect of oxidative stress or chronic inflammation on telomere attrition.10 However, adjustment for these risk factors does not attenuate the association between shorter telomere length and CAD,15 18 suggesting that the relationship does not simply reflect the effect of these risk factors on telomere attrition.
TELOMERE LENGTH IN VASCULAR TISSUES
There is a strong correlation in telomere length between different tissues of the same person.3 29 Therefore, the shorter telomeres seen in the leucocytes in patients with CAD are probably also representative of the situation in the vessel wall. More direct evidence comes from studies that have shown shorter telomeres in coronary endothelial cells from patients with CAD than in subjects without CAD.30 Indeed, probably as a consequence of increased cell turnover, telomere shortening may even be exaggerated at sites prone to atherosclerosis such as bifurcations31 and in cells within the atherosclerotic plaque.32 Similarly, endomyocardial biopsies of subjects with dilated cardiomyopathy display an increased level of cellular senescence and cell death associated with a 39% reduction of average telomere length compared with normal hearts.33
THE TELOMERE HYPOTHESIS
Possibly, the association of shorter telomeres with CAD and other cardiovascular disease simply reflects the greater cell turnover (both of white cells in the circulation and vascular cells) that is known to occur in these conditions and has no direct role in pathogenesis. However, several lines of evidence suggest that the association may be more fundamental:
-
Cellular senescence, of both the endothelium34 and smooth muscle cells,32 is an important and early feature of the atherosclerotic plaques. This is accompanied by changes in gene expression (eg, increased intracellular adhesion molecule-1 (ICAM-1) and decreased levels of nitric oxide synthase (eNOS)) that are implicated in atherogenesis.35
-
Importantly, some of these changes in expression can be directly related to telomere biology. When telomere shortening and dysfunction was induced in human vascular endothelial cells by inhibition of the telomere-associated protein, TRF2, using a mutant lacking the TRF2-binding domain, phenotypic changes characteristic of senescence occurred and the cells exhibited increased ICAM-1 expression and decreased endothelial eNOS activity. Conversely, introduction of the catalytic subunit of telomerase significantly extended the lifespan of the cells, reversed the changes in gene expression, and inhibited the functional alterations associated with senescence.35
-
Similarly, suppressing TRF2 function in cultured cardiac myocytes provoked telomere erosion and apoptosis, while exogenous TRF2 protected against damage from oxidative stress.36
-
Finally, telomerase knockout mice develop shorter telomeres with increasing number of generations and show evidence of cardiac dysfunction.37
These observations suggest that telomeres could play a primary role in CAD and other cardiovascular diseases through the impact of telomere length on the progression to cellular senescence (fig 2). This hypothesis has the potential to integrate different strands in the known aetiology of CAD. It could, for instance, at least partly explain the familial risk of CAD. Rather than specific genes, a more global property of DNA (telomere length) could be the inherited component that transmits the genetic risk. The risk associated with several conventional risk factors for CAD such as hypertension, smoking and diabetes could also be partly mediated through increased telomere attrition mediated by oxidative stress. More speculatively, telomere shortening through increased replicative stress could even play a role in the association between decreased body weight at birth, greater catch-up growth and greater risk of CAD in adulthood.38 Equally importantly, since the various factors that affect telomere length will vary from one person to the next, the telomere hypothesis provides the ability to explain, at least partly, the interindividual variation in susceptibility to, and age of onset of, CAD.

{kind=link}
{kind=link}
There are many aspects of the telomere hypothesis of CAD that require further information and refinement. However, like any good hypothesis it lends itself to testing. For example, if inheritance of shorter telomeres partly explained the genetic basis of CAD, then one would predict that offspring of patients with premature CAD, who are healthy but at higher risk because of their family history, would have shorter telomeres than offspring from families without such a history. Similarly, increased rates of telomere attrition should be associated with greater subsequent risk of CAD. Continuing studies are testing these possibilities.
SUMMARY AND CONCLUSIONS
In the past few years, several studies have shown an association between shorter white cell telomeres and increased risk of CAD. A better understanding of the nature of this association has the potential to offer new insights into the genetic aetiology and pathogenesis of CAD and partly explain the interindividual variation in risk of occurrence and age of onset of CAD. Whether telomere length/biology provides a new therapeutic target for CAD remains to be seen.
Acknowledgments
NJS holds a British Heart Foundation Chair of Cardiology. PvdH is a supported by the Innovational Research Incentives Scheme programme of the Netherlands Organisation for Scientific Research (NWO VENI, grant 916.76.170) and is a research fellow of the Netherlands Heart Foundation (grant 2006T003).
REFERENCES
Footnotes
-
Competing interests: None.