
Abstract
Increased amyloid-β precursor protein (AβPP) and amyloid-β (Aβ) accumulation appear to be upstream steps in the pathogenesis of sporadic inclusion-body myositis (s-IBM). BACE1, participating in Aβ production is also increased in s-IBM muscle fibers. Nogo-B and Nogo-A belong to a family of integral membrane reticulons, and Nogo-B binding to BACE1 blocks BACE1 access to AβPP, decreasing Aβ production. We studied Nogo-B and Nogo-A in s-IBM muscle and in our IBM muscle culture models, based on AβPP-overexpression or ER-stress-induction in cultured human muscle fibers (CHMFs). We report that: (1) in biopsied s-IBM fibers, Nogo-B is increased, accumulates in aggregates, is immuno-co-localized with BACE1, and binds to BACE1; Nogo-A is undetectable. (2) In CHMFs, (a) AβPP overexpression increases Nogo-B, Nogo-A, and BACE1, (b) ER stress increases BACE1 but decreases Nogo-B and Nogo-A, (c) Nogo-B and Nogo-A associate with BACE1. Accordingly, two novel mechanisms, AβPP overexpression and ER stress, are involved in Nogo-B and Nogo-A expression in human muscle. We propose that in s-IBM muscle the Nogo-B increase may represent an attempt by muscle fiber to decrease Aβ production. However, the increase of Nogo-B seems insufficient because Aβ continues to accumulate and the disease progresses. We propose that manipulations, which increase Nogo-B in s-IBM muscle might offer a new therapeutic opportunity.
Similar content being viewed by others

Introduction
Sporadic inclusion-body myositis (s-IBM) is the most common, relentlessly progressive muscle disease of older persons. It leads to severe muscle wasting and disability, and there is no persistently successful treatment [14]. Both, degeneration of muscle fibers and mononuclear cell inflammation are components of s-IBM muscle biopsy pathology [reviewed in 9, 13]. Degeneration of s-IBM muscle fibers includes vacuolization, accompanied by intra-muscle-fiber accumulations of congophilic, ubiquitinated, multi-protein aggregates, which include amyloid-β (Aβ) and paired helical filaments (PHFs) containing phosphorylated tau (p-tau) [8, 9]. An intriguing aspect is that the molecular-morphologic phenotype of s-IBM muscle is similar to Alzheimer-disease (AD) brain [8, 9] including, in addition to accumulated Aβ and p-tau, accumulations of several other “Alzheimer-characteristic” proteins, markers of oxidative stress, and mitochondrial abnormalities [8, 9]. Endoplasmic reticulum stress and proteasome inhibition were also recently shown to be components of the s-IBM pathogenesis [15, 37]. Along with accumulated Aβ, there are increased transcription and accumulation of amyloid-β precursor protein (AβPP) [3, 28]. BACE1 (β-site of the AβPP cleaving enzyme) [23] and components of γ-secretase—presenilin 1 and nicastrin [38]—are also increased [7, 34–36] and are known to participate in abnormal processing of AβPP and Aβ production. Several lines of evidence based on the IBM human muscle culture model and various transgenic mouse models [16, 20, 21, 31], strongly suggest that intra-muscle-fiber accumulation of AβPP and of its proteolytic fragment Aβ plays a key upstream toxic role in s-IBM pathogenesis [5, 6, 8, 9, 15, 24, 41, 43]. Accordingly, we postulate that methods reducing Aβ accumulation in s-IBM muscle fibers could benefit s-IBM patients.
In vitro Nogo-B and RTN3 interact with BACE1 to block BACE1 access to AβPP resulting in decreased Aβ production [18]. Nogo, also known as reticulon 4 (RTN4), belongs to the reticulon (RTN) family of integral membrane proteins [32, 44]. All RTNs, including members of the Nogo family, share a common C-terminal domain but differ in the length of their N-terminal domains [32, 44]. Nogo-A (RTN4A), Nogo-B (RTN4B) and Nogo-C (RTN4C) are the three main isoforms of RTN4 [44]. They are encoded by the same gene, and their variants, generated by alternative splicing or alternative promoter usage, are differentially distributed in various tissues [25, 32, 44]. RTNs are especially abundant in the endoplasmic reticulum [32, 44]. Nogo-B has also been reported in cell surface caveolae/lipid rafts [1]. Nogo-A has been reported to participate in several neurodegenerative disorders [reviewed in 44], and recently it was shown accumulated in the Aβ-containing senile plaques of Alzheimer-disease brains [17].
We examined Nogo-B, Nogo-A and RTN3 in s-IBM muscle fibers by immunoblotting and immunomorphologic techniques, and evaluated whether they physically associate with BACE1. To explore the possible pathogenic mechanisms involved in Nogo regulation in s-IBM, we utilized our established experimentally-induced IBM culture human muscle models, based either on overexpression of AβPP through an adenovirus mediated AβPP gene transfer, or by induction of ER stress [5, 6, 24, 26, 27].
Materials and methods
Muscle biopsies
Studies were performed on diagnostic muscle biopsies obtained with informed consent from 37 patients with the following diagnoses: 14 s-IBM; 4 polymyositis; 2 dermatomyositis; 3 vacuolated non-IBM myopathies (including 2 acid-maltase deficiency patients and 1 vacuolar myopathy of unknown origin); 4 peripheral neuropathies; and 10 normal muscles (no detectable neuromuscular disease). All s-IBM biopsies met the diagnostic criteria detailed in [8].
Immunoblots
Immunoblots of s-IBM and control muscle biopsies were performed as previously described [15, 26, 37, 40]. In brief, 20 μm-thick frozen muscle sections were collected at −25°C and rapidly homogenized on ice in RIPA buffer and protease inhibitor cocktail (Roche Diagnostic GmBH, Mannheim, Germany). Twenty microgram of total muscle protein were electrophoresed in NuPage 3–8% Tris–acetate gel and 1X Tris–acetate SDS running buffer. They were then transferred to a nitrocellulose membrane and incubated with a primary antibody overnight at 4°C. The following well-characterized antibodies against Nogo were used: (a) rabbit polyclonal antibody R461, recognizing Nogo-A/B on immunoblots [18]; (b) goat polyclonal antibody N-18, also recognizing Nogo-A/B [1, 17]; and (c) rabbit polyclonal antibody H-300, recognizing an epitope exclusively present on the N-terminal of Nogo-A [30]. Further, a rabbit polyclonal antibody R454, recognizing RTN3 [18], was also used in preliminary experiments. BACE1 was studied with a mouse monoclonal antibody, clone 61–3E7 [18], and a rabbit polyclonal antibody. Table 1 lists all the antibodies, their dilutions and sources.
After incubation in a primary antibody, membranes were washed and incubated with an appropriate species-specific secondary antibody conjugated to HRP. The blots were developed using the enhanced chemiluminescence system (Amersham Bioscience, Piscataway, NJ, USA). Protein loading was evaluated by actin bands visualized with a mouse monoclonal anti-actin antibody, diluted 1:2,000 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Omission of a primary antibody was the control for specificity of the reactions, and these always produced negative result.
The immunoreactivity was quantified by densitometric analysis using the Kodak Gel Logic 440 imaging system (Eastman Kodak Company, Rochester, NY, USA). Intensity of the band of interest was calculated in relation to intensity of the actin band.
Statistical analysis
The statistical significance was determined by Student’s t test. The significance level was set at P < 0.05. All sets of data are presented as means ± SEM.
Light-microscopic immunocytochemistry
Immunofluorescence was performed on 10-μm-thick transverse sections of fresh-frozen muscle biopsies of all the diseases and controls, as described [3, 15, 26, 37, 40], using the same antibodies that were used for the immunoblots. Double-immunofluorescence utilized goat polyclonal antibody against Nogo-A/B in combination with one of the following: (a) mouse monoclonal antibody against BACE1, or (b) rabbit polyclonal antibody against caveolin-1. To block non-specific binding of an antibody to Fc receptors, all sections were preincubated with 10% normal goat or donkey serum [15, 26, 37, 40]. Omission of the primary antibody or its replacement with non-immune sera or irrelevant antibody was the control for staining specificity, and these always produced negative results.
Gold-immuno-electronmicroscopy
Single- and double-labeled gold immuno-electron-microscopy were performed on 10-μm unfixed frozen sections of s-IBM biopsies adhered to the bottom of 35-mm Petri dishes, as described [3, 15, 26, 37, 40]. Two different species-specific secondary antibodies, conjugated to either 12 or 6 nm gold particles (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), were used in this study. The sections were then processed for EM as previously described [3, 15, 26, 37, 40].
Combined immunoprecipitation–immunoblotting
To evaluate whether in s-IBM muscle fibers (a) Nogo physically associates with BACE1, and (b) BACE1 physically associates with AβPP, we performed the combined immunoprecipitation–immunoblot technique, as described [15, 37, 40, 41]. In brief, 100 μg of total muscle protein were immunoprecipitated in precipitation-buffer containing 5 μg of IgG antibody against BACE1. The immunoprecipitated complex, containing IgG antibody along with its bound target antigen and all proteins bound to that antigen, was pulled down using Protein G Sepharose 4 Fast Flow (Amersham) during 4 h of incubation at 4°C. The solution was then centrifuged for 5 min (16,000×g at 4°C) and the supernatant removed. The precipitated sepharose immunocomplexes were washed three times with the precipitation buffer by centrifuging 5 min each (16,000×g at 4°C). Immunoprecipitates were electrophoresed and immunoprobed with either goat polyclonal anti-Nogo antibody or mouse monoclonal 6E10 antibody against AβPP/Aβ, each followed by an appropriate secondary antibody. To confirm specificity of the physical association identified by the immunoprecipitation–immunoblot reaction, primary antibodies were omitted from either the immunoprecipitation or immunoprobing. The same technique was performed to evaluate whether Nogo physically associates with AβPP.
Cultured human muscle fibers
Primary cultures of normal human muscle were established, as we have described [2], from satellite cells of portions of diagnostic muscle biopsies from patients who, after all tests were performed, were considered free of muscle disease. We established 11 culture sets, each from satellite cells derived from a different muscle biopsy. All experimental and control conditions were studied on sister cultures in the same culture set. Not all studies were performed on every set. Twenty days after myoblasts fused, a 3 Kb 751 AβPP-cDNA, in either sense or anti-sense orientation, was transferred into well-differentiated myotubes using a replication-deficient adenovirus (RDAV) vector at 0.3 × 108 pfu/ml culture medium, as detailed [5, 6, 24]. At the same time, non-AβPP-transduced control sister-cultures were treated for 24 h with either of two ER-stress inducers, Tunicamycin, an N-glycosylation inhibitor (4 μg/ml), or Thapsigargin, an inhibitor of ER calcium-ATPase (300 nM) (both inhibitors from Sigma Co, St Louis, MO, USA) [10, 22, 26]. In our previous studies, both inhibitors successfully induced ER stress in CHMFs [26, 27]. Four days after AβPP gene transfer, or 24 h after treatment with an ER-stress inducer, cultures were processed for light-microscopic immunocytochemistry, immunoblot and immunoprecipitation studies of Nogo and BACE1, as described [15, 26, 41, 43].
Results
Muscle biopsies
Nogo-B and BACE1 are increased in s-IBM muscle fibers
Antibodies R461 and N-18, which were previously characterized as recognizing both Nogo-A and Nogo-B [1, 17, 18], showed a strongly increased level of 46 kDa Nogo-B in s-IBM as compared to controls (Fig. 1a). Densitometric quantification of immunoblots performed on 7 s-IBM and 7 control muscle biopsies revealed that Nogo-B was increased 2.5-fold (P = 0.025) in s-IBM biopsies compared to controls (Fig. 1b). Those antibodies R461 and N-18 should also recognize Nogo-A; however, Nogo-A was not expressed in either normal-control or s-IBM muscle biopsies (Fig. 1a). (In our studies, similarly to previous reports [17, 30], Nogo-A has molecular weight of approximately 180 kDa, which may be due to Nogo-A glycosylation.) Another Nogo-A specific antibody, H-300 (Santa Cruz) also failed to detect any Nogo-A in these samples; a weak band corresponding to 80 kDa was non-specific (Fig. 1a). We found that all three of these antibodies did recognize Nogo-A in a control lysate from human medulloblastoma cells (TE671) that express Nogo-A (Fig. 1a), and also in muscle biopsies from patients with peripheral neuropathy and ALS [42, not shown here]. Together, our results showed that Nogo-B levels are significantly increased in s-IBM while Nogo-A is not detectable. The level of RTN3, another important inhibitor of BACE1 [18], was low in both s-IBM and control biopsies, and there was no discernable difference between the two groups (data not shown). BACE1, which we have previously shown increased and migrating at 70 kDa in s-IBM muscle fibers [34, 36], was, when quantified in the current studies, found increased 3.5-fold (P = 0.007) in s-IBM as compared to control muscle biopsies (Fig. 1c, d).

Immunoblots of Nogo-B, Nogo-A and BACE1 of muscle homogenates of control (C) and s-IBM muscle biopsies. a Demonstrates that a 46 kDa band of Nogo-B detected with antibodies R461 and N-18 was significantly stronger in s-IBM than in control biopsies; however a 180 kDa band corresponding to Nogo-A was not demonstrable with antibodies R461 and N-18, which is considered to recognize both Nogo-A and -B. (This was confirmed with an antibody H-300, which specifically recognizes Nogo-A. Nogo-A was not demonstrable in either control or s-IBM muscle biopsies with this antibody). TE671 indicates immunoblots of an extract of human medulloblastoma cells that express both Nogo-A and -B. In b immunoreactivity was quantified by densitometric analysis using the Kodak Gel Logic 440 imaging system (Eastman Kodak Company, Rochester, NY, USA). Intensity of the band of interest was calculated in relation to the actin band Immunoblot in c and its densitometric analysis in d demonstrate that BACE1 is significantly increased in s-IBM as compared to controls. (Details for a–d in the text, * indicates P < 0.05)
Detection of protein aggregates containing Nogo-B and BACE1 in s-IBM
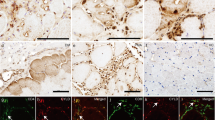
We used immunohistochemistry to morphologically localize Nogo-B in s-IBM muscle fibers. In all of the s-IBM muscle biopsies, 60–70% of the vacuolated muscle fibers contained, mainly in their non-vacuolated cytoplasm, numerous well-defined, plaque-like, punctuate or elongated, aggregates immunoreactive with antibodies R461 and N-18 (Fig. 2a, c, d, f). As shown above by our immunoblots, these two antibodies in s-IBM muscle recognize exclusively Nogo-B, suggesting that the morphologically evident aggregates contain Nogo-B. In addition, in all s-IBM muscle biopsies, 20–30% of the non-vacuolated fibers (on a given cross-section) contained similar aggregates (Fig. 2b). Nogo-B immunoreactive aggregates also contained BACE1 (Fig. 2d, e). As both BACE1 and Nogo-B can be localized in lipid rafts, we found with an antibody against caveolin-1, that caveolin-1 was associated with some of the Nogo-B aggregates (Fig. 2f, g). These results appeared specific because the antibody H-300, which specifically recognizes Nogo-A, and did not detect any specific band in s-IBM muscle biopsies on immunoblots, did not label any aggregates. In contrast to the staining of Nogo-B in s-IBM biopsies, none of the normal or disease-control biopsies had muscle fibers containing Nogo-B immunoreactive aggregates (data not shown). While Nogo-A was negative in s-IBM muscle fibers, it was, as we have recently reported [42], strongly and diffusely immunoreactive in (a) regenerating muscle fibers in dermatomyositis and polymyositis, and (b) in denervated muscle fibers in amyotrophic lateral sclerosis and peripheral neuropathies (not shown).

Immunofluorescence of Nogo-B in s-IBM muscle fibers. a–c Single-label immunofluorescence illustrates strongly-immunoreactive, various-sized aggregates of Nogo-B. d–g Double-label immunofluorescence—the aggregates immunoreactive for Nogo-B (d) are also immunoreactive for BACE1 (e), and aggregates immunoreactive for Nogo-B (f) are also immunoreactive for caveolin-1 (g). a–c ×1,100, d–g ×2,000
Nogo-B and BACE1 ultrastructurally co-localize within s-IBM aggregates
To determine the ultrastructural localization of Nogo-B in the aggregates, we performed immuno-electronmicroscopic examination of s-IBM muscle fibers. Nogo-B was detected on or very close to the 6–10 nm amyloid-like fibrils (Fig. 3a), but was more associated with the cytoplasmic amorphous material (Fig. 3b). Double-labeling experiments showed that BACE1 was co-localized with Nogo-B on 6–10 nm filaments and on the amorphous material (Fig. 3c). Both Nogo-B and BACE1 were also found in the lipid raft structures that were labeled by the antibody against caveolin-1 (Fig. 3d,e). Therefore, Nogo-B immunoreactive aggregates appear to contain BACE1.

Single (a, b) and double-label (c, d, e) gold-immuno-electronmicroscopy of s-IBM muscle fibers. a, b Nogo-B (12 nm gold particles) is localized on 6–10 nm filaments (a) and a large amorphous inclusion (b); on the right of b is a normal myofibrillar structure that does not contain immunoreactive Nogo-B. c Nogo-B (12 nm gold particles) and BACE1 (6 nm gold particles) are associated with 6–10 nm amyloid-like fibrils and with amorphous-floccular material. d both Nogo-B (12 nm gold particles) and BACE1 (6 nm gold particles) are immunolocalized in the caveolae structures, and on 6–10 nm filaments. e Double-labeling of a caveola with antibodies against Nogo-B (12 nm gold particles) and caveolin-1 (6 nm gold particles). a–d ×53,000; c ×68,000
Physical interaction between BACE1 and Nogo-B
Since the ultrastructural results suggest that BACE1 and Nogo-B co-exist in s-IBM aggregates, we next tested whether, within s-IBM muscle fibers, they physically interact as previously reported in the human brain [18]. Immunoprecipitation with an antibody against BACE1 followed by immunoblotting with antibody N-18, which in s-IBM muscle biopsies recognized only Nogo-B, revealed a 46-kDa band corresponding to Nogo-B. Omission of a primary antibody against BACE1 from the immunoprecipitation reaction (denoted by ×) or in the immunoblot (denoted by #) failed to detect Nogo-B (Fig. 4a). The co-immunoprecipitation experiments also demonstrated that in s-IBM muscle biopsies BACE1 interacted with AβPP (Fig. 4b). However, there was no physical association of Nogo-B with AβPP/Aβ (Fig. 4c). Together, these studies indicate that Nogo-B and BACE1 are present in aggregates where they appear to have interacted.

a, b Immunoprecipitations of s-IBM biopsies with anti-BACE1 antibody, followed by immunoprobing with an antibody against Nogo A/B in a, and with antibody 6E10 against AβPP/Aβ in b. A strong band of the appropriate molecular weight of 46 kDa corresponding to Nogo-B is visible in a. In b, a strong band of 130 kDa corresponds to AβPP. × = omission of the antibody against BACE1 from the immunoprecipitation solution; # = omission of the antibody against Nogo-B in a or omission of the antibody against AβPP/Aβ (6E10) in b in the immunoblotting—these omissions resulted in negative reactions. Together the data indicate there is a physical interaction between Nogo-B and BACE1 in s-IBM muscle fibers. c Illustrates that there is no interaction between AβPP/Aβ and Nogo-B
Cultured human muscle fibers
Increased expression of AβPP enhances Nogo-B and BACE1 protein levels
To determine what might be causing increased expression of Nogo-B and BACE1 in s-IBM muscle fibers, we utilized our IBM human culture model, which exhibits several abnormalities present in s-IBM muscle fibers [6, 15, 24, 41, 43]. Since increased production and accumulation of AβPP and Aβ, as well as ER-stress, seem to be important components of the s-IBM pathogenesis, in this study we: (a) engineered cultured human muscle fibers (CHMFs) to overexpress AβPP through an adenoviral AβPP cDNA transfer, and (b) separately induced ER-stress in CHMFs.
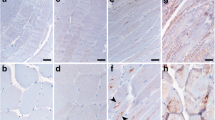
Densitometric analysis of immunoblots demonstrated that in the AβPP-overexpressing CHMFs (lane AβPP+) compared to controls (Fig. 5a, b) a 46 kDa band corresponding to Nogo-B was increased 1.3-fold (P = 0.018) and Nogo-A was increased 1.7 fold (P = 0.038). AβPP overexpression in CHMFs also caused 1.9-fold (P = 0.022) increase of BACE1 as compared to controls (Fig. 5c, d). These effects of AβPP overexpression were specific because overexpressing AβPP in antisense orientation in CHMFs did not cause increase of Nogo-B, Nogo-A, or BACE1 (Fig. 5e–h).

Immunoblots performed with cultured human muscle fibers (CHMFs). a Immunoblots of control—C, Tg- and Tm-exposed, and AβPP-overexpressing CHMFs, and their densitometric analyses (b), show that Nogo-A and -B are significantly increased in AβPP-overexpressing CHMFs, but both are decreased in CHMFs in which ER-stress was induced by Tg or Tm treatment. c, d BACE1 is significantly increased due to AβPP overexpression, or treatment with Tg or Tm. e–h Illustrates that overexpressed antisense-AβPP does not influence Nogo-A or-B, or BACE1 in CHMFs. a–h * indicates P < 0.05)
Contrarily to AβPP-overexpression, treatment of CHMFs with two ER-stress inducers, thapsigargin (Tg) or tunicamycin (Tm), led to decreased level of Nogo-B (1.3-fold [P = 0.019] by thapsigargin, and 1.2-fold [P = 0.04] by tunicamycin), and of Nogo-A (1.4-fold [P = 0.001] by thapsigargin and 1.5-fold [P = 0.001] by tunicamycin) (Fig. 5a, b). However, both ER-stress inducers increased BACE1 levels—1.4-fold (P = 0.027) after thapsigargin treatment and 1.7-fold (P = 0.011) after tunicamycin treatment (Fig. 5c, d).
By immunofluorescence, in normal, non-AβPP-overexpressing CHMFs, Nogo and BACE1 were weakly and diffusely immunoreactive (Fig. 6a, b), whereas in the AβPP-overexpressing (AβPP+) cultures their immunoreactivities were stronger and occasionally in aggregates (Fig. 6c, d). (Because CHMFs express both Nogo-B and Nogo-A, and the antibodies N-18 and R461 that we utilized for immunostaining recognize both Nogo-A and Nogo-B, we use term “Nogo” to refer to those two stainings.) In the aggregates, Nogo and BACE1 co-localized (Fig. 6c, d). In the CHMFs, interactions of BACE1 with Nogo-B and Nogo-A (Fig. 7a) or with AβPP (Fig. 7b) were further confirmed through our co-immunoprecipitation experiments.

a–d Double-label immunofluorescence of “Nogo” (a, c) with BACE1 (b, d), in the control (a, b) and the AβPP-overexpressing (c, d) CHMFs. This illustrates the co-localization of Nogo and BACE1, and the stronger immunostaining of both due to the AβPP overexpression. An upper fiber in c and d contains two large aggregates containing both Nogo and BACE1. All ×1,200

Immunoprecipitation-immunoblotting illustrates that both Nogo-B and -A associate with BACE1 (a), and BACE1 associates with AβPP/Aβ (c). # = omission of the primary antibody from the immunoblotting reaction
Discussion
In s-IBM muscle fibers, we demonstrated by immunoblot analysis, using well-characterized anti-Nogo antibodies, that Nogo-B is significantly increased and Nogo-A is virtually undetectable. Our immunocytochemical studies showed that in s-IBM muscle fibers Nogo-B was abnormally accumulated multifocally in the form of aggregates that also contained BACE1 and caveolin-1. By immuno-electronmicroscopy, both Nogo-B and BACE1 were associated with the same ultrastructural components, namely, amorphous material, 6–10 nm amyloid-like fibrils and caveolae/lipid-rafts-like structures, which also contained caveolin-1. Previous studies in other cells have also shown that Nogo-B and BACE1 are present in the caveolae/lipid-rafts structures, and processing of AβPP leading to Aβ generation is considered to occur in those structures as well [1, 12, 23, 33].
The two-reticulon family members, RTN3 and Nogo-B, were previously shown to bind BACE1 in human brain and in HEK-293 cells [18]. This binding was recently shown to occur through the C-terminal domain common to all reticulons and the C-terminal domain of BACE1 [19]. This binding inhibits association of BACE1 with AβPP and decreases generation of Aβ-40 and Aβ-42 [18, 19]. Accordingly, within s-IBM muscle fibers, the increased Nogo-B and its association with BACE1, might reflect an attempt by those fibers to decrease Aβ production caused by BACE1-mediated abnormal processing of AβPP.
In our cultured human muscle fibers, AβPP overexpression increased both Nogo-B and Nogo-A, as well as the intracellular amount of BACE1. We propose that the increased BACE1 is in response to an increased demand to process the excessive AβPP, resulting in increased Aβ production. Although the mechanism(s) of increased Nogo-B and -A, in this system needs to be clarified, we suggest that they might be to inhibit (at least partially) BACE1 binding to AβPP, thereby decreasing production of Aβ and protecting the muscle fiber from the Aβ detrimental influence. Since both Nogo-B and Nogo-A bind to BACE1 in our CHMFs, we propose that both Nogo isoforms could protect a muscle fiber from an abnormal accumulation of Aβ; however, in the s-IBM biopsied muscle fibers, only Nogo-B seems to be playing this putative role. Because Nogo-A is increased in regenerating muscle fibers [42], it may have additional roles in those young fibers. One possibility might be to help manage the increased AβPP known to occur in regenerating muscle fibers in vivo [4, 29]—this putative benefit of Nogo-A could be deficient in s-IBM muscle fibers, since they have diminished regenerative capabilities, and we could not demonstrate Nogo-A in them. Nogo-A is predominantly expressed in the central nervous system, where it is present mainly in oligodendrocytes [11, 39]. A recent study demonstrated that Nogo-A, but not Nogo-B, is increased in hippocampal neurons in Alzheimer disease (AD) brain, and it also co-localizes with Aβ deposits in extracellular senile plaques [17]. Similarly to our present studies, Nogo-A did not physically associated with AβPP/Aβ in AD brain [17]; whether Nogo-A physically associated with BACE1 in AD brain was not reported [17].
In our cultured human muscle fibers, ER stress decreased Nogo-A/B and increased BACE1 levels as compared to the unstressed control. Analogously, we suggest that in s-IBM muscle fibers, continuous ER stress [27, 37] might be reducing the intracellular ratio of Nogo-B to BACE1 by decreasing Nogo-B and increasing BACE1, resulting in Aβ continuing to accumulate and exert its cytotoxic influence.
Conclusions
We report abnormally increased Nogo-B and its multi-focal accumulation within s-IBM muscle fibers. We experimentally demonstrate that two different mechanisms, namely AβPP overexpression and endoplasmic reticulum stress, are differentially involved in regulation of Nogo-B, Nogo-A and BACE1 in cultured human muscle fibers. Since both AβPP overexpression and ER-stress are important pathologic aspects of the s-IBM muscle fibers, the same regulatory mechanism may operate in them. If so, manipulations toward increasing Nogo-B and/or interfering with pathways inducing ER-stress might provide new therapeutic opportunities for s-IBM patients.
References
Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC (2004) A new role for Nogo as a regulator of vascular remodeling. Nat Med 10:382–388
Askanas V, Engel WK (1992) Cultured normal and genetically abnormal human muscle, in the handbook of clinical neurology. In: Rowland LP, Di Mauro S (eds) Myopathies, vol 18. North Holland, Amsterdam, pp 85–116
Askanas V, Alvarez RB, Engel WK (1993) β-amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol 34:551–560
Askanas V, Sarkozi E, Bilak M, Engel WK (1994) Expression of β-amyloid precursor protein, prion and acetylcholine receptor and their mRNAs in human muscle fibers regenerating in vivo. Brain Pathol 4:322
Askanas V, McFerrin J, Baque S, Alvarez RB, Sarkozi E, Engel WK (1996) Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc Natl Acad Sci USA 93:1314–1319
Askanas V, McFerrin J, Alvarez RB, Baque S, Engel WK (1997) βAPP gene transfer into cultured human muscle induces inclusion-body myositis aspects. Neuroreport 8:2155–2158
Askanas V, Engel WK, Yang CC, Alvarez RB, Lee VM, Wisniewski T (1998) Light and electron microscopic immunolocalization of presenilin 1 in abnormal muscle fibers of patients with sporadic inclusion-body myositis and autosomal-recessive inclusion-body myopathy. Am J Pathol 152:889–895
Askanas V, Engel WK (2001) Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol 60:1–14
Askanas V, Engel WK (2006) Inclusion-body myositis: a myodegenerative conformational disorder associated with Aβ, protein misfolding, and proteasome inhibition. Neurology 66:S39–S48
Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35:395–416
Buss A, Sellhaus B, Wolmsley A, Noth J, Schwab ME, Brook GA (2005) Expression pattern of NOGO-A protein in the human nervous system. Acta Neuropathol (Berl) 110:113–119
Cordy JM, Hooper NM, Turner AJ (2006) The involvement of lipid rafts in Alzheimer’s disease. Mol Membr Biol 23:111–122
Dalakas MC (2006) Inflammatory, immune, and viral aspects of inclusion-body myositis. Neurology 66:S33–S38
Engel WK, Askanas V (2006) Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology 66:S20–S29
Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW, Askanas V (2005) Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol 167:517–526
Fukuchi K, Pham D, Hart M, Li L, Lindsey JR (1998) Amyloid-beta deposition in skeletal muscle of transgenic mice: possible model of inclusion body myopathy. Am J Pathol 153:1687–1693
Gil V, Nicolas O, Mingorance A, Urena JM, Tang BL, Hirata T, Saez-Valero J, Ferrer I, Soriano E, del Rio JA (2006) Nogo-A expression in the human hippocampus in normal aging and in Alzheimer disease. J Neuropathol Exp Neurol 65:433–444
He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R (2004) Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat Med 10:959–965
He W, Hu X, Shi Q, Zhou X, Lu Y, Fisher C, Yan R (2006) Mapping of interaction mediating binding between BACE1 and RTN/Nogo proteins. J Mol Biol 363:625–634
Jin LW, Hearn MG, Ogburn CE, Dang N, Nochlin D, Ladiges WC, Martin GM (1998) Transgenic mice over-expressing the C-99 fragment of betaPP with an alpha-secretase site mutation develop a myopathy similar to human inclusion body myositis. Am J Pathol 153:1679–1686
Kitazawa M, Green KN, Caccamo A, LaFerla FM (2006) Genetically augmenting Abeta42 levels in skeletal muscle exacerbates inclusion body myositis-like pathology and motor deficits in transgenic mice. Am J Pathol 168:1986–1997
Lee AS (2005) The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35:373–381
Marlow L, Cain M, Pappolla MA, Sambamurti K (2003) Beta-secretase processing of the Alzheimer’s amyloid protein precursor (APP). J Mol Neurosci 20:233–239
McFerrin J, Engel WK, Askanas V (1998) Impaired innervation of cultured human muscle overexpressing Amyloid-betaPP experimentally and genetically: relevance to inclusion-body myopathies. Neuroreport 9:3201–3205
Ng CE, Tang BL (2002) Nogos and the Nogo-66 receptor: factors inhibiting CNS neuron regeneration. J Neurosci Res 67:559–565
Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V (2006) Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem 96:1491–1499
Nogalska A, Wojcik S, King Engel W, McFerrin J, Askanas V (2007) Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: Relevance to inclusion body myositis. Exp Neurol 204:610–618
Sarkozi E, Askanas V, Johnson SA, Engel WK, Alvarez RB (1993) beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport 4:815–818
Sarkozi E, Askanas V, Johnson SA, McFerrin J, Engel WK (1994) Expression of beta-amyloid precursor protein gene is developmentally regulated in human muscle fibers in vivo and in vitro. Exp Neurol 128:27–33
Satoh J, Onoue H, Arima K, Yamamura T (2005) Nogo-A and nogo receptor expression in demyelinating lesions of multiple sclerosis. J Neuropathol Exp Neurol 64:129–138
Sugarman MC, Yamasaki TR, Oddo S, Echegoyen JC, Murphy MP, Golde TE, Jannatipour M, Leissring MA, LaFerla FM (2002) Inclusion body myositis-like phenotype induced by transgenic overexpression of beta APP in skeletal muscle. Proc Natl Acad Sci USA 99:6334–6339
Teng FY, Ling BM, Tang BL (2004) Inter- and intracellular interactions of Nogo: new findings and hypothesis. J Neurochem 89:801–806
Tun H, Marlow L, Pinnix I, Kinsey R, Sambamurti K (2002) Lipid rafts play an important role in A-beta biogenesis by regulating the beta-secretase pathway. J Mol Neurosci 19:31–35
Vattemi G, Engel WK, McFerrin J, Buxbaum JD, Pastorino L, Askanas V (2001) Presence of BACE1 and BACE2 in muscle fibres of patients with sporadic inclusion-body myositis. Lancet 358:1962–1964
Vattemi G, Kefi M, Engel WK, Askanas V (2003) Nicastrin, a novel protein participating in amyloid-beta production, is overexpressed in sporadic inclusion-body myositis muscle. Neurology 60:A315
Vattemi G, Engel WK, McFerrin J, Pastorino L, Buxbaum JD, Askanas V (2003) BACE1 and BACE2 in pathologic and normal human muscle. Exp Neurol 179:150–158
Vattemi G, Engel WK, McFerrin J, Askanas V (2004) Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol 164:1–7
Vetrivel KS, Thinakaran G (2006) Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 66:S69–73
Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM (2002) Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci 22:5505–5515
Wojcik S, Engel WK, McFerrin J, Askanas V (2005) Myostatin is increased and complexes with amyloid-beta within sporadic inclusion-body myositis muscle fibers. Acta Neuropathol (Berl) 110:173–177
Wojcik S, Engel WK, McFerrin J, Paciello O, Askanas V (2006) AbetaPP-overexpression and proteasome inhibition increase alphaB-crystallin in cultured human muscle: relevance to inclusion-body myositis. Neuromuscul Disord 16:839–844
Wojcik S, Engel WK, Askanas V (2006) Increased expression of Nogo-A in ALS muscle biopsies is not unique for this disease. Acta Myologica 25:116–118
Wojcik S, Nogalska A, McFerrin J, Engel WK, Oledzka G, Askanas V (2007) Myostatin precursor protein is increased and associates with amyloid-β precursor protein in inclusion-body myositis culture model. Neuropathol Appl Neurobiol 33:238–242
Yan R, Shi Q, Hu X, Zhou X (2006) Reticulon proteins: emerging players in neurodegenerative diseases. Cell Mol Life Sci 63:877–889
Acknowledgments
Slawomir Wojcik is on leave from the Department of Anatomy and Neurobiology, Medical University of Gdansk, Gdansk, Poland. Maggie Baburyan provided excellent technical assistance in electronmicroscopy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by grants (to VA) from the National Institutes of Health (AG 16768 Merit Award), the Muscular Dystrophy Association, and The Myositis Association (to VA), and the Helen Lewis Research Fund.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wojcik, S., Engel, W.K., Yan, R. et al. NOGO is increased and binds to BACE1 in sporadic inclusion-body myositis and in AβPP-overexpressing cultured human muscle fibers. Acta Neuropathol 114, 517–526 (2007). https://doi.org/10.1007/s00401-007-0281-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0281-y