Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: a population based cohort study
BMJ 2010; 340 doi: https://doi.org/10.1136/bmj.c693 (Published 09 February 2010) Cite this as: BMJ 2010;340:c693
- Catherine M Kelly, medical oncology fellow 15,
- David N Juurlink, division head, clinical pharmacology123457,
- Tara Gomes, epidemiologist7,
- Minh Duong-Hua, analyst6,
- Kathleen I Pritchard, professor1235,
- Peter C Austin, senior statistician57,
- Lawrence F Paszat, senior scientist12357
- 1Department of Medicine, Sunnybrook Health Sciences Centre, Toronto, Canada
- 2Sunnybrook Research Institute, Toronto,
- 3Department of Medicine, University of Toronto, Toronto
- 4Department of Pediatrics, University of Toronto
- 5Department of Health Policy, Management, and Evaluation, University of Toronto
- 6Canadian Institute for Health Information, Toronto
- 7The Institute for Clinical Evaluative Sciences, Ontario, Canada
- Correspondence to: D Juurlink, Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada M4N 3M5 dnj{at}ices.on.ca
- Accepted 1 February 2010
Abstract
Objective To characterise whether some selective serotonin reuptake inhibitor (SSRI) antidepressants reduce tamoxifen’s effectiveness by inhibiting its bioactivation by cytochrome P450 2D6 (CYP2D6).
Design Population based cohort study.
Participants Women living in Ontario aged 66 years or older treated with tamoxifen for breast cancer between 1993 and 2005 who had overlapping treatment with a single SSRI.
Main outcome measures Risk of death from breast cancer after completion of tamoxifen treatment, as a function of the proportion of time on tamoxifen during which each SSRI had been co-prescribed.
Results Of 2430 women treated with tamoxifen and a single SSRI, 374 (15.4%) died of breast cancer during follow-up (mean follow-up 2.38 years, SD 2.59). After adjustment for age, duration of tamoxifen treatment, and other potential confounders, absolute increases of 25%, 50%, and 75% in the proportion of time on tamoxifen with overlapping use of paroxetine (an irreversible inhibitor of CYP2D6) were associated with 24%, 54%, and 91% increases in the risk of death from breast cancer, respectively (P<0.05 for each comparison). By contrast, no such risk was seen with other antidepressants. We estimate that use of paroxetine for 41% of tamoxifen treatment (the median overlap in our sample) would result in one additional breast cancer death within five years of cessation of tamoxifen for every 19.7 (95% confidence interval 12.5 to 46.3) patients so treated; the risk with more extensive overlap would be greater.
Conclusion Paroxetine use during tamoxifen treatment is associated with an increased risk of death from breast cancer, supporting the hypothesis that paroxetine can reduce or abolish the benefit of tamoxifen in women with breast cancer.
Introduction
Breast cancer is the most commonly diagnosed cancer in women worldwide, and it is estimated that 1.5 million new cases will be diagnosed in 2010.1 Tamoxifen is a selective oestrogen receptor modulator that has been used for the treatment of breast cancer for over three decades.2 In women with early stage oestrogen receptor positive breast cancer, tamoxifen reduces the risk of recurrence by about half and the risk of breast cancer death by about a third. These benefits are largely independent of chemotherapy, age, progesterone receptor status, or other tumour characteristics.3
Tamoxifen is a prodrug that is metabolised by the hepatic cytochrome P450 enzyme system to the active metabolites 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen (endoxifen).4 5 Both metabolites have an affinity for the oestrogen receptor that is 100-fold higher than the parent compound; however, endoxifen is considered the most important metabolite because its plasma concentrations are several times higher than those of 4-hydroxytamoxifen.6 7 Conversion of tamoxifen to endoxifen is catalysed predominantly by cytochrome P450 isoenzyme 2D6 (CYP2D6).4 7 This enzyme is highly polymorphic,8 and in some studies loss-of-function variants are associated with lower endoxifen concentrations,5 an increased risk of breast cancer relapse and a shorter time to recurrence during tamoxifen therapy.9 10 11 12 13 14 Consequently, therapy with drugs that inhibit CYP2D6 may reduce the clinical benefit of tamoxifen by interfering with its bioactivation, particularly when these drugs are used for an extended period. Indeed, in patients who receive tamoxifen in combination with a CYP2D6 inhibitor, endoxifen concentrations vary inversely with the degree of CYP2D6 inhibition.5 15 16
Up to 25% of patients with breast cancer experience a depressive disorder.17 Newer antidepressants are widely used in women with breast cancer for treatment of depression and are prescribed for tamoxifen related hot flashes and various other indications.18 19 20 21 This practice is particularly relevant in the context of tamoxifen therapy because selective serotonin reuptake inhibitor (SSRI) antidepressants inhibit CYP2D6 to varying degrees. For example, paroxetine is an exceptionally potent CYP2D6 inhibitor, and is the only SSRI that exhibits mechanism based (“suicide”) inhibition, resulting in irreversible loss of enzyme function until new CYP2D6 is synthesised.22 23 24
Whether antidepressant related inhibition of CYP2D6 is associated with adverse outcomes in patients receiving tamoxifen is unknown. To explore this possibility, we linked prescribing records with detailed clinical data from a large population based cancer registry and other population based healthcare datasets to explore the clinical consequences of the potential interaction between SSRIs and tamoxifen.
Methods
Setting and design
We did a population based retrospective cohort study among female residents of Ontario, Canada, who were aged 66 years or older and who started tamoxifen treatment between 1 January 1993 and 31 December 2005. These individuals have universal access to health care, including hospital care, doctor’s services, and prescription drug coverage. The study was approved by the research ethics board of Sunnybrook Health Sciences Centre, Toronto, Canada.
Data sources
We analysed the computerised prescription records of the Ontario Public Drug Benefit Program, which contains comprehensive records of prescription medications dispensed to all Ontario residents aged 65 or older. We identified women with breast cancer using the Ontario Cancer Registry, a population based tumour registry that contains pathology reports, hospital discharge abstracts, and death certificates of patients with a diagnosis of cancer. Hospital admissions were identified using the Canadian Institute for Health Information Discharge Abstract database, which contains information on hospital visits, including detailed clinical and demographic information on all hospital admissions. Doctors’ services were identified using the Ontario Health Insurance Plan database, and we obtained basic demographic information, including date of death, from the Registered Persons database. We estimated socioeconomic status by linking residential postal codes with Statistics Canada population census data. These datasets were linked using an encrypted version of each patient’s 10 digit health card number to ensure anonymity, and they are regularly used to study drug safety, including the clinical consequences of drug interactions.25 26 27 28 29 30
Design and analysis
We identified a cohort of women starting tamoxifen treatment, beginning with the first tamoxifen prescription after each patient’s 66th birthday. All patients were newly treated with tamoxifen (defined as no tamoxifen prescription in the preceding year), and had a diagnosis of breast cancer in the Ontario Cancer Registry (International Classification of Disease version 9 (ICD-9) codes 174.0-174.9). We did not include patients during their first year of eligibility for prescription drug coverage (age 65) to avoid incomplete medication records.
For each patient, we determined the total duration of tamoxifen treatment by aggregating the total days supplied for all tamoxifen prescriptions. We restricted our analysis to women whose tamoxifen use encompassed at least 80% of the total number of days between the first and last tamoxifen prescriptions. In addition, we restricted our analysis to women co-prescribed a single SSRI antidepressant (paroxetine, fluoxetine, sertraline, citalopram, or fluvoxamine) during tamoxifen treatment. We also included venlafaxine, which inhibits serotonin reuptake as well as norepinephrine reuptake at higher doses.31 We did not study duloxetine or escitalopram because they were not insured benefits of the provincial formulary during the study period.
For analytical purposes, we defined the index date as the date on which tamoxifen was last dispensed, plus an additional 60 days. This allowed us to completely ascertain the total duration of tamoxifen treatment and the extent to which co-prescription of potentially interacting medications occurred during the course of treatment. Moreover, it allowed us to adequately characterise the mortality consequences of the drug interaction between SSRIs and tamoxifen, which are delayed and largely occur after the completion of tamoxifen treatment, in contrast with most other clinically important drug interactions, which usually have more immediate clinical consequences.29 30 32
In the primary analysis, patients were followed up from the index date until death from breast cancer or the end of the study period (31 December 2007), whichever occurred first. A secondary analysis considered death from any cause, including breast cancer. For each patient, we quantified the duration of tamoxifen treatment and the proportion of this time that was characterised by concomitant SSRI treatment. Because we expected that the consequences of CYP2D6 inhibition during tamoxifen therapy would be delayed, we excluded women who switched from one SSRI to another while taking tamoxifen. Consequently, all women in our analysis were 66 years or older, newly treated with tamoxifen for breast cancer, and treated with a single SSRI antidepressant during tamoxifen treatment.
The primary outcome was death from breast cancer (ICD9 codes 174.0 to 174.9 and ICD10 codes C50.0 to 50.9). We used Cox proportional hazards regression to examine the effect of SSRI co-prescribing on survival following cessation of tamoxifen. In each analysis, we expressed exposure as the proportion of time on tamoxifen that was characterised by overlapping SSRI treatment.
For each SSRI, we estimated hazard ratios and 95% confidence intervals associated with increasing proportions of co-prescribing of that same drug with tamoxifen, thereby restricting our analyses to within-drug comparisons. Consequently, the analysis explored the relation between a continuous measure (proportion of overlap between each SSRI and tamoxifen) and breast cancer mortality, without the need for a reference group. The regression model was adjusted for age, year that tamoxifen was started, duration of tamoxifen treatment, timing of tamoxifen in relation to date of breast cancer diagnosis (within one year of diagnosis or thereafter), socioeconomic status, comorbidity in the year before completion of tamoxifen treatment,33 and co-prescription of other CYP2D6 inhibitors (bupropion, quinidine, thioridazine, amiodarone, cimetidine, or chloroquine) during tamoxifen treatment. The proportional hazards assumption was verified using Schoenfeld residuals.34 All analyses were done with SAS version 9.1 (SAS Institute, Cary N.C.) and used a two-tailed type 1 error rate of 0.05 as the threshold for statistical significance.
Results
We identified 24 430 women aged 66 years and older who started tamoxifen treatment during the 13 year study period (appendix 1). Of these, 7489 (30.6%) received at least one antidepressant during tamoxifen treatment. After excluding those treated with no SSRI or with multiple SSRIs, those with poor adherence to tamoxifen, and those with unknown cause of death, the primary analysis included 2430 women (table 1)⇓. The median age in the year before starting tamoxifen was 74 years (interquartile range (IQR) 70-79).
Characteristics by antidepressant group
Most patients (n=2025; 83.3%) started tamoxifen treatment within one year of breast cancer diagnosis. The median duration of tamoxifen treatment was 4.0 years (IQR 2.2-5.0), ranging from 3.2 years (1.4-4.8) in women who also received fluoxetine to 4.2 years (2.5-5.1) among women who also received citalopram (appendix 2). Paroxetine was the most commonly prescribed SSRI (n=630; 25.9%) followed by sertraline (n=541; 22.3%), citalopram (n=467; 19.2%), venlafaxine (n=365; 15.0%) fluoxetine (n=253; 10.4%) and fluvoxamine (n=174; 7.2%). Overall, 735 patients (30.2%) received at least one other non-SSRI antidepressant, including 445 (18.3%) who received a cyclic antidepressant, 209 (8.6%) who received other antidepressants, and 81 (3.3%) who received both. The distribution of these patients was relatively similar across the various SSRI groups (appendix 3).
Main analyses
In total, 1074 women (44.2%) died by the end of follow-up (mean follow-up 2.38 years, SD 2.59), including 374 (15.4%) in whom breast cancer was recorded as the cause of death. In the primary analysis, we found an increased risk of death from breast cancer among women who received paroxetine, an irreversible CYP2D6 inhibitor, in combination with tamoxifen. After adjusting for potential confounders, absolute increases of 25%, 50%, and 75% in the proportion of time on tamoxifen that overlapped with use of paroxetine were associated with relative increases of 24%, 54%, and 91% in the risk of death from breast cancer, respectively (table 2⇓, fig 1⇓). In contrast, we found no increased risk of breast cancer mortality associated with exposure to the other SSRIs during tamoxifen treatment. We noted a non-significant trend towards reduced breast cancer mortality among venlafaxine users (table 2⇓), which might reflect the common practice of using venlafaxine for tamoxifen-related hot flashes,35 a potential predictor of better outcomes in women receiving tamoxifen.36

Fig 1 Risk of breast cancer mortality associated with increasing proportions of antidepressant use during tamoxifen treatment
{kind=link}
Antidepressant exposure and risk of death from breast cancer in women receiving tamoxifen
To test the robustness of our conclusions, we replicated our analyses using death from any cause (including breast cancer) as the outcome of interest (n=1074). After adjusting for potential confounders, we found that absolute increases of 25%, 50%, and 75% in paroxetine exposure during tamoxifen treatment were associated with relative increases of 13%, 28% and 46%, respectively, in the risk of death from any cause (table 3⇓, fig 2⇓). By contrast, we found no such increased risk in all-cause mortality associated with exposure to the other SSRIs in women receiving tamoxifen for breast cancer. Finally, we conducted an additional analysis including 226 women whose cause of death was unknown (total n=1300). This analysis again yielded consistent results, showing an increased risk of death only with paroxetine.
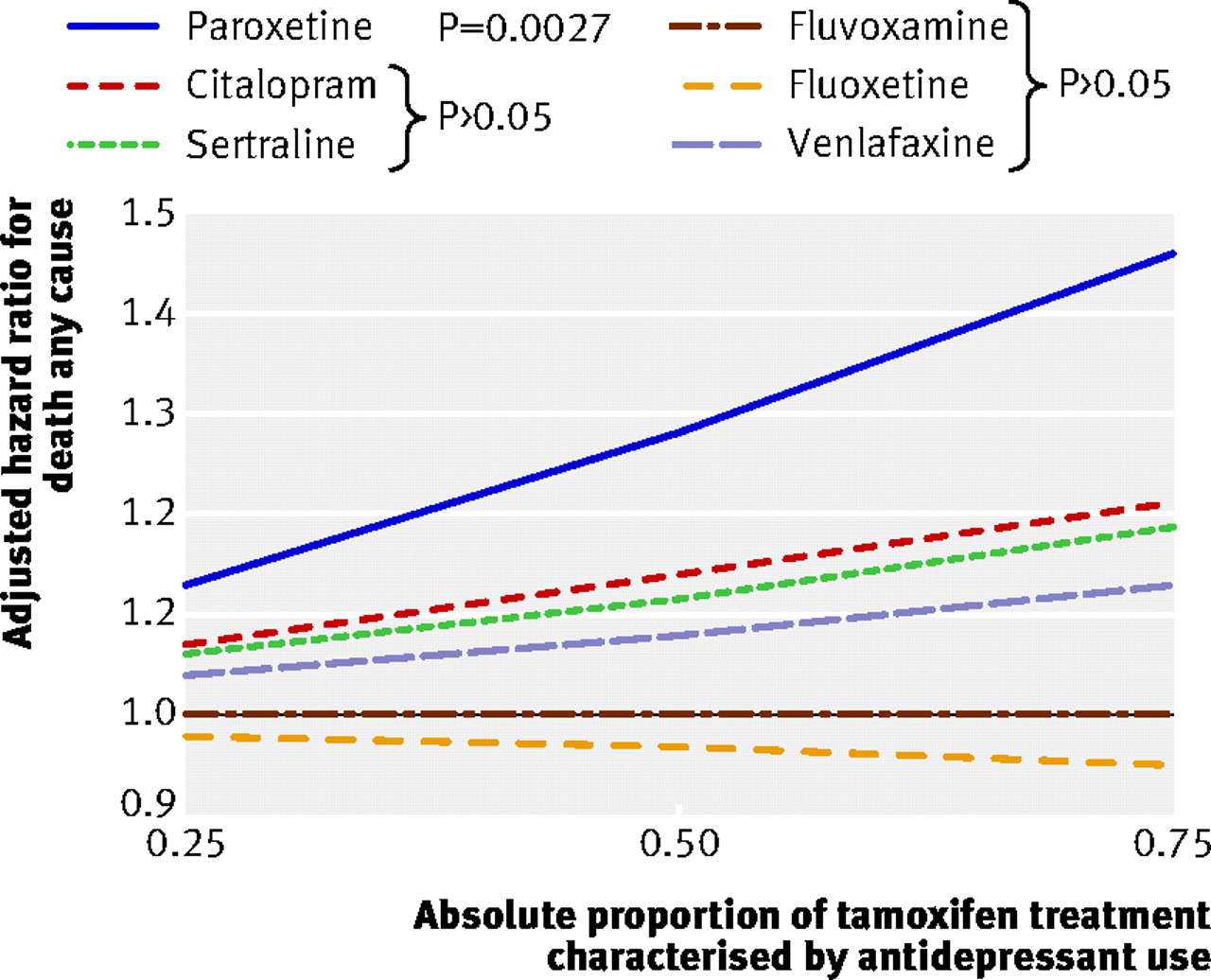
Fig 2 All-cause mortality associated with increasing proportions of antidepressant use during tamoxifen therapy
{kind=link}
Antidepressant exposure and risk of death from any cause in women receiving tamoxifen
Estimate of absolute risks
To better characterise the absolute risks imparted by use of paroxetine with tamoxifen, we generated absolute risk estimates from our primary analysis using methods described elsewhere37 and applied previously.38 These estimates were obtained using a five year period from the end of tamoxifen treatment, and their reciprocal is the number needed to treat to harm (NNTh).
Compared with patients with minimal (1%) overlap of paroxetine with tamoxifen, use of paroxetine for 41% of tamoxifen treatment (the median overlap in our sample 8% to 80%) would result in one additional breast cancer death at five years for every 19.7 (95% confidence interval 12.5 to 46.3) women so treated. Similarly, compared with patients with a 1% overlap, we estimate that use of paroxetine for the entire duration of tamoxifen (100% overlap) would result in an additional death for every 6.9 (95% confidence interval 4.3 to 18.6) patients so treated.
Discussion
Using population based healthcare data, we found that women with breast cancer who received paroxetine in combination with tamoxifen were at increased risk for death from breast cancer and death from any cause. The increased risk was directly related to the extent of co-prescribing and is consistent with the hypothesis that irreversible CYP2D6 inhibition by paroxetine can reduce or abolish the survival advantage conferred by long term tamoxifen therapy in patients with breast cancer. We found no such risk with other antidepressants. Our findings are consistent with an emerging body of literature indicating the critical role of CYP2D6 in the metabolic activation 4 5 15 and clinical effectiveness of tamoxifen.9 10 11 12 13 14
Implications for clinical practice
Our findings have major implications for clinical practice, particularly in light of the frequency of combination therapy. The prevalence of depression in women with early breast cancer is roughly twice that of the general female population and is particularly high around the time of diagnosis.39 In our study, 30% of women who started tamoxifen treatment also received antidepressants, and paroxetine was the most commonly used SSRI. These patients may also take SSRIs for other indications; up to 80% of women treated with tamoxifen experience hot flashes,40 and clinical trials have shown the efficacy of SSRIs for their treatment.20
Strengths of the study
Our findings differ from previous research reporting no significant association between SSRI treatment and adverse outcome in women receiving tamoxifen. Insufficient statistical power, use of a case control design, and assessment of a single SSRI with weak CYP2D6-inhibiting activity18 41 42 43 might have hampered the ability of these studies to detect clinically important differences in outcome. In the context of drug interactions, case control studies are better suited to the study of short term risks resulting from drug interactions rather than long term risks.25 29 30 32 44
Limitations of the study
Some limitations of our study merit emphasis. We could not ascertain the indication for antidepressant treatment, but our finding of an increased mortality risk with paroxetine has strong biological plausibility and is not readily explained by selection bias. Some women taking tamoxifen may have been prescribed newer antidepressants for treatment of tamoxifen related hot flashes,20 which have been associated with better response to treatment.36 These observations may underlie the trend towards lower breast cancer mortality observed with venlafaxine, which exhibits minimal CYP2D6 inhibition and is commonly used for hot flashes.45 46 47 Although other SSRIs may also be used for hot flashes, confounding by indication is an unlikely explanation for our remaining observations, as this would tend to result in negative associations between increasing drug overlap and breast cancer mortality.
We did not have information on breast cancer stage, which is an important predictor of outcome. However, because we conducted a within-SSRI analysis, this is unlikely to bias our findings. There is no reason why women with more advanced breast cancer should be preferentially prescribed paroxetine rather than other antidepressants. We cannot exclude the possibility that individuals who received longer durations of paroxetine while taking tamoxifen had more severe disease, although this seems clinically implausible.21 48
About 7% of individuals exhibit no functional CYP2D6 activity 7 and are therefore unable to convert tamoxifen to endoxifen. These individuals may have fewer tamoxifen associated hot flashes, and may have better adherence to tamoxifen but a poorer response to the drug.49 Although we do not have genotype information on our study participants, the inclusion in our analysis of patients with loss-of-function polymorphisms will tend to minimise the clinical consequences of drug induced CYP2D6 inhibition, and can only attenuate the ability of our analysis to discriminate among SSRIs.
The finding of an increased risk of death from any cause in women co-prescribed paroxetine has at least two contributing explanations. First, breast cancer was the most common cause of death in these patients, and an association between paroxetine use and total mortality was therefore expected. Second, some deaths not specifically ascribed to breast cancer might have reflected remote effects of the disease (such as pulmonary embolism or cardiac tamponade) or the disease itself, particularly when no other cause of death was recorded. Importantly, these limitations apply to all antidepressants, and cannot explain the differential mortality risk observed with paroxetine treatment.
Although the degree to which various SSRIs inhibit CYP2D6 differs among studies, there is consensus that both fluoxetine and its metabolite are strong inhibitors of CYP2D6.50 51 However, we found no association between increasing use of fluoxetine and death from breast cancer among women taking tamoxifen. The reasons for this are unclear, but might reflect the relatively small number of women exposed to fluoxetine in our study sample. Our results should not be viewed as evidence that fluoxetine can be safely used in combination with tamoxifen. Similarly, we cannot exclude the possibility that insufficient sample size explains the non-significant mortality results with other SSRIs.
Conclusion
In conclusion, our findings indicate that the choice of antidepressant can significantly affect survival in women receiving tamoxifen for breast cancer. This observation is consistent with the critical role of CYP2D6 in the metabolic activation of tamoxifen, and highlights a drug interaction that is extremely common, widely underappreciated and uniformly avoidable. Tamoxifen is a crucial element of treatment for patients with hormone receptor positive breast cancer regardless of age or breast cancer stage. When co-prescription of tamoxifen with an antidepressant is necessary, preference should be given to antidepressants that show little or no inhibition of CYP2D6.
What is already known on this topic
Tamoxifen is important in the endocrine treatment of breast cancer and is a prodrug converted by cytochrome P450 2D6 (CYP2D6) to its active metabolite endoxifen.
Selective serotonin inhibitor (SSRI) antidepressants are widely prescribed to women with breast cancer taking tamoxifen, but inhibit CYP2D6 to varying degrees and may affect tamoxifen’s effectiveness.
What this study adds
Use of paroxetine (a potent, irreversible CYP2D6 inhibitor) during tamoxifen treatment is associated with an increased subsequent risk of death due to breast cancer in a fashion that correlates with the duration of combined use.
We estimate that treatment with paroxetine for 41% of tamoxifen therapy (the median in our study) could result in one additional breast cancer death at five years for every 20 women so treated.
Notes
Cite this as: BMJ 2010;340:c693
Footnotes
We thank David Henry, John Horn, and Steven Shumak for comments on an earlier draft of this manuscript, and we thank Ashif Kachra for assistance with manuscript preparation.
Participants: CMK (guarantor): study concept, design, data extraction, analysis, manuscript writing. DNJ: study concept, design, analysis, supervision, manuscript writing. TG: study design, analysis of data. MD-H: data extraction and analysis. KIP: study design, supervision. PCA: study design and analysis of data. LFP: study concept, design, analysis, supervision. CMK and DNJ had full access to the data (including statistical reports and tables) in the study. CMK takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding: CMK was supported by a fellowship award from the Sunnybrook Hospital Foundation. DNJ was supported by a New Investigator award from the Canadian Institutes of Health Research. PCA is supported by a Career Investigator award from the Heart and Stroke Foundation of Ontario. LFP is supported by a Clinician Scientist award from the Ontario Ministry of Health and Long-Term Care. This research was supported by the Institute for Clinical Evaluative Sciences (ICES), which is funded by an annual grant from the Ontario Ministry of Health and Long-Term Care. The study was supported in part by the Ontario Drug Policy Research Network, funded by the Ontario Drug Innovation Fund. The funder had no role in the design, analysis, conduct or reporting of the study. The opinions, results and conclusions reported in this paper are those of the authors and are independent from the funding sources. No endorsement by ICES or the Ontario MOHLTC is intended or should be inferred.
Competing interests: All authors have completed the Unified Competing Interest form at www.icmje.org_disclosure.pdf (available on request from the corresponding author. During the past three years KIP has been a consultant for Sanofi-Aventis, AstraZeneca, Roche, Pfizer, Ortho-Biotech, YM Biosciences, Novartis, Abraxis, Amgen, and GlaxoSmithKline (GSK). KIP has received research funding either directly through per case funding for studies, or indirectly through the National Cancer Institute of Canada Clinical Trials Group, contracted with pharmaceutical companies including AstraZeneca, YM Biosciences, Bristol Myers, Squibb, Sanofi-Aventis, Amgen, Ortho-Biotech, Pfizer, Novartis, GlaxoSmithKline, and Ortho Biotech. KIP has received honorariums or been part of speaker’s bureaux from Sanofi-Aventis, AstraZeneca, Pfizer, Roche, YM Biosciences, and Novartis. KIP has given paid expert testimony for Sanofi-Aventis, AstraZeneca, and GlaxoSmithKline. KIP has been a member of Advisory Committees for Sanofi-Aventis, AstraZeneca, Ortho-Biotech, Roche, Pfizer, Novartis, YM Biosciences, and GlaxoSmithKline. All other authors declare (1) no support from any company for the submitted work; (2) no relationships with any companies that might have an interest in the submitted work in the previous 3 years; (3) their spouses, partners, or children have no financial relationships that may be relevant to the submitted work; and (4) no non-financial interests that may be relevant to the submitted work.
Ethical approval: The study was approved by the research ethics board of Sunnybrook Health Sciences Centre, Toronto, Canada.
Data sharing: No additional data available
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.