Sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of epidemiological surveillance data for 1970-96
BMJ 1997; 315 doi: https://doi.org/10.1136/bmj.315.7105.389 (Published 16 August 1997) Cite this as: BMJ 1997;315:389
- S N Cousens, senior lecturer (s.cousens{at}lshtm.ac.uk)a,
- M Zeidler, research registrarb,
- T F Esmonde, research registrarb,
- R De Silva, research registrarb,
- J W Wilesmith, head of epidemiology departmentc,
- P G Smith, professor of tropical epidemiology and department heada,
- R G Will, consultant neurologistb
- a Department of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, London WC1E 7HT
- b National Creutzfeldt-Jakob Disease Surveillance Unit, Western General Hospital, Edinburgh EH4 2XU
- c Central Veterinary Laboratory, Addlestone, Surrey KT15 3NB
- Correspondence to: Mr Cousens
- Accepted 19 March 1997
Abstract
Objective: To identify changes in the occurrence of Creutzfeldt-Jakob disease that might be related to the epidemic of bovine spongiform encephalopathy.
Design: Epidemiological surveillance of the United Kingdom population for Creutzfeldt-Jakob disease based on (a) referral of suspected cases by neurologists, neuropathologists, and neurophysiologists and (b) death certificates.
Setting: England and Wales during 1970-84, and whole of the United Kingdom during 1985-96.
Subjects: All 662 patients identified as sporadic cases of Creutzfeldt-Jakob disease.
Main outcome measures: Age distribution of patients, age specific time trends of disease, occupational exposure to cattle, potential exposure to causative agent of bovine spongiform encephalopathy.
Results: During 1970-96 there was an increase in the number of sporadic cases of Creutzfeldt-Jakob disease recorded yearly in England and Wales. The greatest increase was among people aged over 70. There was a statistically significant excess of cases among dairy farm workers and their spouses and among people at increased risk of contact with live cattle infected with bovine spongiform encephalopathy. During 1994-6 there were six deaths from sporadic Creutzfeldt-Jakob disease in the United Kingdom in patients aged under 30.
Conclusions: The increase in the incidence of sporadic Creutzfeldt-Jakob disease and the high incidence in dairy farmers in the United Kingdom may be unrelated to bovine spongiform encephalopathy. The most striking change in the pattern of Creutzfeldt-Jakob disease in the United Kingdom after the epidemic of bovine spongiform encephalopathy is provided by the incidence in a group of exceptionally young patients with a consistent and unusual neuropathological profile. The outcome of mouse transmission studies and the future incidence of the disease in the United Kingdom and elsewhere, will be important in judging whether the agent causing bovine spongiform encephalopathy has infected humans.
Key messages
The overall incidence of Creutzfeldt-Jakob disease in the United Kingdom has increased in recent years
The largest increases have been in the oldest age groups and probably represent improved case ascertainment rather than a real change in incidence
The comparatively high incidence of Creutzfeldt-Jakob disease in dairy farmers in the United Kingdom is comparable to that observed in dairy farmers in countries where bovine spongiform encephalopathy is rare or absent
The observation of a group of comparatively young patients with Creutzfeldt-Jakob disease characterised by unusual neuropathological features during 1994-6 remains unexplained
The results of mouse transmission studies and the future incidence of Creutzfeldt-Jakob disease in the United Kingdom and elsewhere will be important in judging whether the causative agent of bovine spongiform encephalopathy has infected humans
Introduction
The United Kingdom National Creutzfeldt-Jakob Disease Surveillance Unit was set up in May 1990 after recognition of the epidemic of bovine spongiform encephalopathy in the late 1980s.1 If bovine spongiform encephalopathy infects humans it was considered most likely to result in disease manifestations similar to those of Creutzfeldt-Jakob disease and that some or all of the following phenomena might be observed: (a) an increase in the overall incidence of Creutzfeldt-Jakob disease in the United Kingdom; (b) an excess of cases in groups most likely to have high exposure to the causative agent of bovine spongiform encephalopathy; (c) a change in the epidemiological pattern of Creutzfeldt-Jakob disease, such as a change in the age distribution; (d) a change in the clinical or neuropathological characteristics of Creutzfeldt-Jakob disease. The identification in 1996 of a group of comparatively young patients with Creutzfeldt-Jakob disease characterised by unusual clinical and neuropathological features2 led the Spongiform Encephalopathy Advisory Committee to advise the United Kingdom government that “the most likely explanation at present is that these cases are linked to exposure to BSE.”3
We present data on age specific changes in the incidence of Creutzfeldt-Jakob disease in the United Kingdom since 1970 and examine the incidence rates of the disease since 1990 in groups who might have been occupationally exposed to the causative agent of bovine spongiform encephalopathy.
Methods
Surveillance of Creutzfeldt-Jakob disease
A series of studies has attempted to identify all cases of Creutzfeldt-Jakob disease occurring in England and Wales since 1970 and in the United Kingdom since 1985. Methods of case ascertainment have been described.2 4 Briefly, cases of Creutzfeldt-Jakob disease occurring in England and Wales during 1970-9 were identified retrospectively at the end of that period5 and prospective surveillance instituted to detect cases during 1980-4.6 Prospective ascertainment of cases, extended to cover the whole of the United Kingdom, was reinstituted in May 1990 and is continuing. Cases occurring in the United Kingdom between 1985 and April 1990 were identified retrospectively.
Case ascertainment has been based throughout on direct notification of cases and on death certificates. Neurologists, neuropathologists, and neurophysiologists are asked to refer suspected cases of Creutzfeldt-Jakob disease, and the Office for National Statistics supplies death certificates coded under rubrics 339.9 and 781.7 (International Classification of Diseases, eighth revision (ICD-8)) and 046.1 and 331.9 (ICD-9). A few cases are referred by other routes.
Since May 1990 suspected cases have been seen, when possible, by a neurologist from the national surveillance unit and a close relative has been interviewed to collect epidemiological information, including the current and previous occupations of the patient.
Case definition
The diagnostic criteria for Creutzfeldt-Jakob disease have been essentially the same since surveillance began in 1970.7 In summary, classification as a definite case requires neuropathological confirmation of spongiform change, or (since 1993) immunocytochemical confirmation of the presence of prion protein, or identification of scrapie associated fibrils on electron microscopy. Currently about 70% of patients with suspected Creutzfeldt-Jakob disease come to necropsy. In the absence of neuropathological investigation patients are classified as probable cases of Creutzfeldt-Jakob disease if they present with progressive dementia, a typical electroencephalogram, and at least two of the following: myoclonus; visual or cerebellar signs; pyramidal or extrapyramidal signs; a kinetic mutism.
Cases are classified as iatrogenic when the patients have a known risk factor for accidental transmission (for example, pituitary hormone recipients). The criteria for inherited Creutzfeldt-Jakob disease have been updated to include information available from genetic analyses. Cases are now classified as inherited if the patients carry a disease specific mutation of the prion protein gene or have a history of probable or definite Creutzfeldt-Jakob disease in a first degree relative. Molecular biological data were available for 53% of cases identified since May 1990. All other cases are classified as sporadic. Probably during 1970-90 roughly half of inherited cases of Creutzfeldt-Jakob disease—that is, one or two a year—may have been classified as sporadic because genetic analyses were not carried out during the period.
Groups with potentially high occupational exposure
We do not know how or indeed if bovine spongiform encephalopathy is transmitted to humans. We identified three routes through which transmission might be hypothesised to occur.
Route 1 might be via contact with ruminant derived meat and bone meal contaminated by bovine spongiform encephalopathy (the likely source of the bovine epidemic).8 Occupational groups identified to be at high risk of this exposure were workers on farms with cattle, pigs, poultry, sheep, or goats (as ruminant derived meat and bone meal may have been fed to these animals) and workers in feed mills producing meat and bone meal.
Route 2 might be via contact with live cattle infected with bovine spongiform encephalopathy. Occupational groups identified to be at high risk of this exposure were workers on farms with cattle, veterinarians, and abattoir workers. Three different populations of farm workers with increasing likelihood of exposure to live infected cattle were identified—namely, workers on all farms with cattle (dairy or beef herds); workers on farms with dairy cattle (the incidence of bovine spongiform encephalopathy has been much higher in dairy herds)9; workers on dairy or beef farms with a confirmed case of bovine spongiform encephalopathy.
Route 3 might be via contact with brain or spinal cord from dead infected cattle. Occupational groups identified to be at high risk of this exposure were veterinarians, abattoir workers, workers in rendering plants, and butchers. Though cattle may be slaughtered on farms by licensed personnel and dressed by farmers, we have no evidence that this is widespread and farmers were therefore excluded from this occupational group.
The numbers of workers on different types of farms during 1990-4 were obtained from the annual agricultural censuses for England and Wales, Northern Ireland, and Scotland (1990-5). Data on the numbers of workers on farms affected by bovine spongiform encephalopathy were obtained by linking agricultural census data with the database on the disease held at the Central Veterinary Laboratory. The number of veterinarians, butchers, and meat cutters in Great Britain was estimated from the 10% sample of the 1991 census by using the recommended conversion factor of 10.16. The number of abattoir workers could not be derived from published census data for Great Britain, but information on these workers was provided by the Ministry of Agriculture, Fisheries and Food.
Data for Northern Ireland on veterinarians and on workers employed in the slaughtering of animals and the production of meat for 1993 were provided by the Department of Economic Development, Belfast. We could not derive the number of workers in rendering plants or feed mills producing meat and bone meal from published census data for Great Britain. However, the numbers of workers in these groups are small compared with the number of farm workers (see below) and their exclusion from calculation of the expected number of cases is therefore of no consequence. (The number of feed mill workers is estimated at about 3000 (United Kingdom Agricultural Supply Trade Association, personal communication, 1996).)
Expected numbers of cases in different occupational groups
Expected numbers of deaths from Creutzfeldt-Jakob disease between 1 May 1990 and 31 December 1996 were calculated by multiplying age specific and sex specific death rates for the United Kingdom population by the estimated numbers of people in the different occupational groups. The age and sex distribution of farm workers was assumed to follow that reported for farm workers in England and Wales in the European Commission's structure survey of 1990. The age and sex distributions of veterinarians and of butchers and meat cutters were assumed to follow those of the wider occupational groups within which they were classified in the 1991 census.
Observed and expected numbers of cases were compared in the assumption that the observed number of cases followed a Poisson distribution.
Results
Between 1 January 1970 and 31 December 1996, 708 definite and probable cases of Creutzfeldt-Jakob disease were identified. Five were in patients alive on 31 December 1996. In all, 540 (76%) cases were classified as definite. Twenty three cases were classified as iatrogenic and 23 as inherited, both of which groups were excluded from analysis. Among the 662 sporadic cases, 15 were of the variant form of the disease.
Distribution of cases by age, sex, and time period
In England and Wales the yearly number of deaths from Creutzfeldt-Jakob disease increased from around 10 at the beginning of the 1970s to around 40 in the 1990s (fig 1). No increase in yearly numbers of deaths was evident for Scotland and Northern Ireland after 1985 (fig 1).
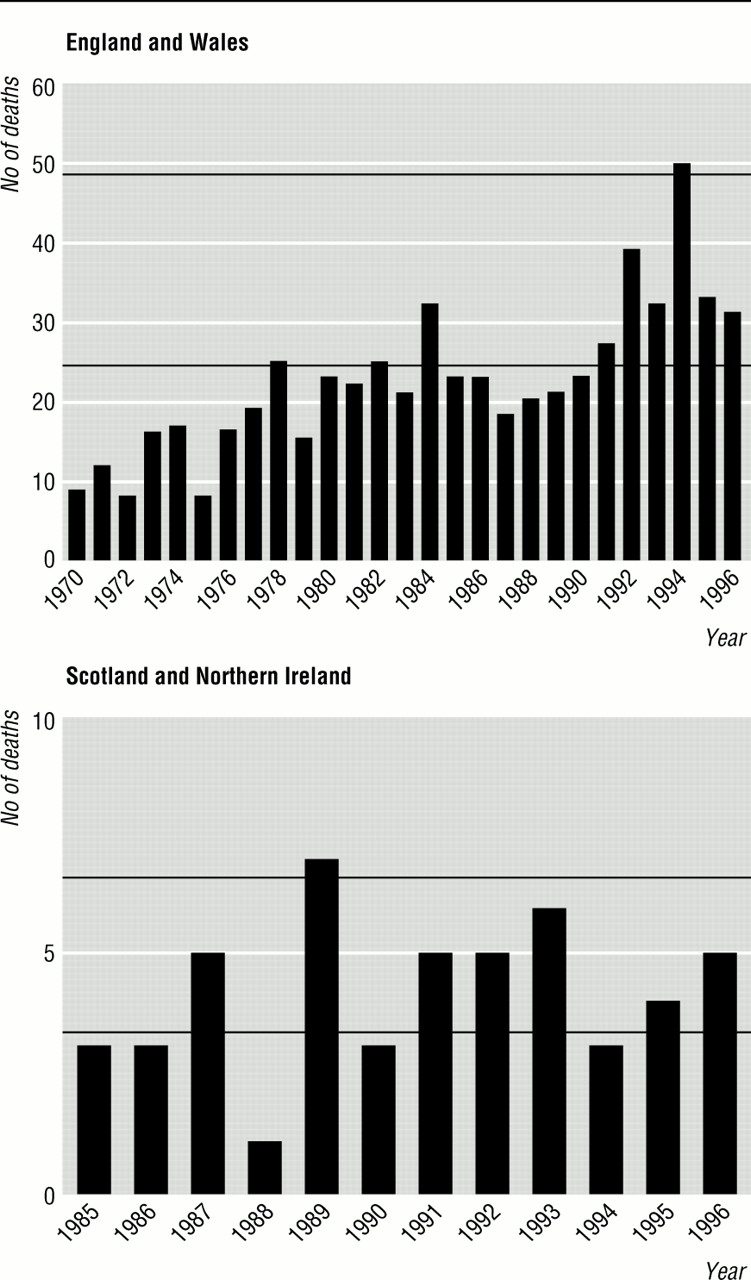
Yearly numbers of deaths from Creutzfeldt-Jakob disease in England and Wales during 1970-96 and in Scotland and Northern Ireland during 1985-96 (data exclude known iatrogenic and inherited cases). Horizontal lines represent numbers of deaths corresponding to yearly death rates of 0.5/million (lower) and 1.0/million (upper)
{kind=link}
Figure 2 shows the average yearly age specific and sex specific death rates over the study period. Below 40 years of age, death rates were extremely low (<0.1/million yearly). Death rates increased substantially in the 50-59 year age group and reached a peak of around 2.0/million yearly in the 60-69 year age group before declining in people aged 70 and over.

Age and sex specific average yearly death rates from Creutzfeldt-Jakob disease in United Kingdom during 1970-96
{kind=link}
Table 1 shows the numbers of deaths in 10 year age groups for each two year period from 1970 to 1996. Mortality from Creutzfeldt-Jakob disease increased substantially over the period in patients aged 70 and over and by successively smaller proportions in those aged 60-69 and 50-59 (fig 3; table 2). The increase in patients aged 40-49 years was not significant (P=0.27). Under the age of 40 numbers of cases were small, but of the seven patients who died under the age of 30, six had onset of the disease after 1 January 1994. All six died in 1995 or 1996.
Numbers of deaths from Creutzfeldt-Jakob disease in 10 year age groups for each two year period in England and Wales (from 1970) and United Kingdom* (from 1985)

Trends in age specific death rates from Creutzfeldt-Jakob disease in England and Wales during 1970-96. For age group 40-49 years death rates for 1972-3 and 1984-5 are not plotted as there were no deaths in this age group in the periods
{kind=link}
Rate of increase in mortality from sporadic Creutzfeldt-Jakob disease by age group
Sporadic disease in groups with potential occupational exposure
Six cases of sporadic Creutzfeldt-Jakob disease (five definite, four in men) were identified from 1 May 1990 in people whose occupation at disease onset was in one of the groups identified above. Four have been the subject of case reports.10 11 12 13 Ages at death in the six cases were 54, 54, 59, 61, 64, and 64 years. All five patients subjected to neuropathological investigation showed changes typical of Creutzfeldt-Jakob disease. None showed the “florid” plaques characteristic of the variant form of the disease.2 The sixth, probable case followed a clinical course typical of sporadic Creutzfeldt-Jakob disease with a characteristic electroencephalogram.
Four of the six patients lived or worked on dairy farms (on three of which there had been confirmed cases of bovine spongiform encephalopathy), and two lived or worked on farms with beef suckler herds (one with a confirmed case of bovine spongiform encephalopathy). Two of the six cases were in spouses of farmers. All patients had lived or worked on farms throughout their working lives. Table 3 shows the distribution of these cases by occupational group. There was a significant excess of cases among animal farm workers (6 observed, 2.4 expected; P=0.03). All these cases were in cattle farmers, four of whom worked on farms with a case of bovine spongiform encephalopathy. This excess of cases was highly significant (4 observed, 0.58 expected; P=0.003). Table 4 shows the distribution of observed and expected cases by hypothesised route of transmission. There was an excess of cases among workers potentially exposed to ruminant derived meat and bone meal and among workers exposed to live cattle infected with bovine spongiform encephalopathy. Two cases among farmers were on farms without a case of bovine spongiform encephalopathy compared with an expected number of 1.78 (P=0.5).
Distribution of expected and observed cases of Creutzfeldt-Jakob disease among occupational groups potentially at increased risk of infection with bovine spongiform encephalopathy
Distribution of expected and observed cases of Creutzfeldt-Jakob disease by hypothesised route of exposure to agent of bovine spongiform encephalopathy
Discussion
In our introduction we identified four changes in the pattern of Creutzfeldt-Jakob disease that might occur if exposure to the bovine agent caused spongiform encephalopathy in humans. Arguably all of these changes may have occurred. There are, however, important caveats that inhibit clear interpretation of our findings.
The number of sporadic cases of Creutzfeldt-Jakob disease recorded in England and Wales has increased. During a period of prospective surveillance before the advent of bovine spongiform encephalopathy (1980-4) the yearly number of deaths from sporadic Creutzfeldt-Jakob disease in England and Wales averaged 24.8. During prospective surveillance after the epidemic of bovine spongiform encephalopathy (1990-6) the yearly number of deaths averaged 33.6. However, similar increases in the incidence of Creutzfeldt-Jakob disease have been observed in countries where the bovine disease is rare or absent (table 5).14 15 These increases may reflect improved case ascertainment rather than real increases in incidence. It has long been suspected that the age distribution of Creutzfeldt-Jakob disease in the United Kingdom (fig 2) and elsewhere—with a decline in incidence in people over 70—might reflect under ascertainment of cases in older age groups.16 In the United Kingdom the greatest increase in incidence has been in people aged over 70. For many diseases case ascertainment is likely to be poorest among old people and best among age groups in which death from any cause is unusual. Thus aside from the recent increase in cases in young people, the age specific time trends suggest that improved ascertainment may explain much or all of the apparent rise in the incidence of Creutzfeldt-Jakob disease since 1970.
Incidence of Creutzfeldt-Jakob disease in countries other than United Kingdom over time
We found significant excesses of cases of Creutzfeldt-Jakob disease among workers on dairy farms and on farms with a confirmed case of bovine spongiform encephalopathy. None of the six patients who might have been occupationally exposed to the agent of bovine spongiform encephalopathy presented with the variant form of Creutzfeldt-Jakob disease, which might indicate a different aetiology from the variant form. However, evidence from iatrogenic cases that route of infection may be a determinant of the clinical and pathological pattern of disease17 means that we cannot exclude entirely the possibility that “occupational” cases and the variant form of the disease arise from exposure to the same agent by different routes. One possible explanation for the apparent excess risk among farmers is that case ascertainment of Creutzfeldt-Jakob disease in the United Kingdom has been better in this group than among other groups because of awareness of a possible link between the bovine and human diseases. Moreover, though the incidence of Creutzfeldt-Jakob disease among dairy farmers in the United Kingdom is higher than in the general population, it does not seem remarkable when compared with the incidence among dairy farmers in other European countries where bovine spongiform encephalopathy is rare or absent (table 6).15 This suggests that dairy farmers may be at increased risk of Creutzfeldt-Jakob disease for reasons other than exposure to the causative agent of the bovine disease. We emphasise also that, though some farm workers seem to be at increased risk of Creutzfeldt-Jakob disease compared with the general population, their absolute risk remains extremely low, the yearly incidence of the disease being well below 10 cases/million.
Incidence rates of Creutzfeldt-Jakob disease in dairy farmers in different European countries
Our analyses are based on occupation at the time of disease onset, as national data are not available on the numbers of people ever employed in particular occupational groups. We therefore excluded from the analyses one patient who had worked in an abattoir for 10 months, six years before disease onset in 1995; one patient who had worked on a farm with dairy and beef herds until retiring in 1991, four years before disease onset in 1995; and one patient with the variant form of the disease who had worked as a butcher between 1985 and 1987. Another case of interest that was similarly excluded was of a man who transported meat and bone meal from an animal feed compounder to farms for many years up to 1991 but who changed his job before onset of Creutzfeldt-Jakob disease in 1993.
Cases with unusual features
During 1994-6, 10 cases of sporadic Creutzfeldt-Jakob disease occurred in the United Kingdom in patients under the age of 30. Based on past experience in the United Kingdom (two cases in the preceding 24 years) and elsewhere2 this is unusual. That nine of these patients together with five others aged 30, 32, 34, 39, and 48 years at onset also exhibited a previously undescribed neuropathological pattern renders this finding even more remarkable and raises important questions. Were these cases really “new” or had similar cases occurred previously without being identified? If these cases were new why did they occur in the United Kingdom in the mid-1990s?
Bruton et al reviewed the Runwell Hospital Brain Archive and identified 19 cases of spongiform encephalopathy, only 11 of which had been diagnosed before death.18 All the patients died before 1985, and none showed the neuropathological changes characteristic of the variant form of Creutzfeldt-Jakob disease. The authors suggested that prion diseases may be more common than supposed. Only about 4% of elderly patients dying with dementia come to necropsy.19 Taken with our finding of a substantial increase over time in the number of cases identified in older age groups, it seems likely that some cases of Creutzfeldt-Jakob disease in this age group have been missed. To maintain the hypothesis that the phenomenon of cases in young people with striking neuropathological changes is not new requires that young people dying of progressive neurological disorders over the past several decades came to necropsy sufficiently rarely for such cases to have been missed entirely. Since 1979 about 46% of patients aged under 45 dying of dementias with which Creutzfeldt-Jakob disease might be confused have come to necropsy, with no evidence that necropsy rates have increased over that time.19 Thus though we cannot exclude the possibility that such cases were missed, it seems to us an unlikely explanation.
If we accept for the present that these young cases are a new phenomenon, why did they appear in the United Kingdom in the mid-1990s? Bovine spongiform encephalopathy has been advanced as the most likely explanation.3 Other workers have asked why in the absence of direct evidence of a link bovine spongiform encephalopathy should be singled out ahead of microwave ovens, high voltage power lines, or organophosphorous sheep dips.20 These exposures are not confined to the United Kingdom and therefore seem less plausible explanations than bovine spongiform encephalopathy. The one case of the variant form of Creutzfeldt-Jakob disease reported in France21 does not compromise the plausibility of the link with bovine spongiform encephalopathy. France has been one of the biggest markets for exported British beef, beef products, and cattle (B Schreuder, J Wilesmith, O Straub, personal communication, 1996).22
One argument against the plausibility of a link between bovine spongiform encephalopathy and the variant form of Creutzfeldt-Jakob disease is that the variant disease has been observed mainly in young people. The lack of cases in older age groups could be due to misdiagnosis, reduced susceptibility, or age related exposure to the agent.2 That the variant form of the disease may have been missed in old people seems plausible but that it would be missed in patients in their late 40s and early 50s seems less so. That age related exposure explains the observed age distribution of the cases is also difficult to sustain, as the link between age and exposure would have to be very strong, and we have identified no exposure related to bovine spongiform encephalopathy that meets this condition.
In summary, the epidemiological data available suggest that the distribution of Creutzfeldt-Jakob disease in the United Kingdom population may be changing. Neuropathological data suggest that a new form of the disease may have appeared which could partly but not wholly explain the epidemiological anomalies observed. There is no direct evidence that links bovine spongiform encephalopathy with these changes, but recent observations accord with this hypothesis.23 24 The results of transmission studies from human Creutzfeldt-Jakob disease cases to mice and the future incidence of Creutzfeldt-Jakob disease in the United Kingdom and elsewhere will be important in judging whether the causative agent of bovine spongiform encephalopathy has infected humans.
Hill et al have recently reported the findings of a Western blot analysis of protease resistant prion protein (PrPRES from five of the six farmers reported here.25 On the basis of fragment size and glycoform ratio of this protein, all five were classified as having type I or type II PrPRES. These are types associated with the classical form of Creutzfeld-Jakob disease and not with the variant form. These authors conclude that their results, in conjunction with the phenotype of these patients, do not support a link with occupational exposure to the agent of bovine spongiform encephalopathy.
Acknowledgments
We thank J Mackenzie for data management; W B Matthews for his foresight in initiating surveillance of Creutzfeldt-Jakob disease in the United Kingdom in 1980 and for subsequent advice; R Knight and R Harries-Jones for invaluable contributions to surveillance of the disease in England and Wales during 1970-84; and James Ironside, Jeanne Bell, Linda McCardle, and Caroline Barrie for neuropathological data. We also thank all neurologists, neuropathologists, and neurophysiologists in Britain, without whose help Creutzfeldt-Jakob disease surveillance would not be possible.
Funding: The Creutzfeldt-Jakob disease surveillance unit is funded by the Department of Health and the Scottish Home and Health Department and supported by the Biotechnology and Biological Sciences Research Council (grant No 15/BS204814).
Conflict of interest: None.